Qsymia Prescribing Information
Package insert / product label
Generic name: phentermine hydrochloride and topiramate
Dosage form: capsule, extended release
Drug class: Anorexiants
Medically reviewed by Drugs.com. Last updated on Dec 13, 2023.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Drug Abuse and Dependence
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
QSYMIA (phentermine and topiramate extended-release capsules), for oral use, CIV
Initial U.S. Approval: 2012
Indications and Usage for Qsymia
QSYMIA is a combination of phentermine, a sympathomimetic amine anorectic, and topiramate, indicated as an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in:
- Adults with an initial body mass index (BMI) of:
- Pediatric patients aged 12 years and older with BMI in the 95 thpercentile or greater standardized for age and sex.
Limitations of Use:
- The effect of QSYMIA on cardiovascular morbidity and mortality has not been established ( 1).
- The safety and effectiveness of QSYMIA in combination with other products intended for weight loss, including prescription and over-the-counter drugs, and herbal preparations, have not been established ( 1).
Qsymia Dosage and Administration
- Take orally once daily in morning. Avoid administration in evening to prevent insomnia ( 2.3).
- Recommended starting dosage is 3.75 mg/23 mg (phentermine mg/topiramate mg) daily for 14 days; then increase to 7.5 mg/46 mg daily ( 2.3).
- Escalate dosage based on weight loss in adults or BMI reduction in pediatric patients. See the Full Prescribing Information for details regarding discontinuation or dosage escalation ( 2.3).
- Gradually discontinue 15 mg/92 mg dosage to prevent possible seizure ( 2.4).
- Do not exceed 7.5 mg/46 mg dosage for patients with moderate or severe renal impairment or patients with moderate hepatic impairment ( 2.5, 2.6).
Dosage Forms and Strengths
Contraindications
Warnings and Precautions
- Embryo-Fetal Toxicity: Can cause fetal harm . In patients who can become pregnant, a negative pregnancy test is recommended before initiating QSYMIA and monthly during therapy; advise use of effective contraception. QSYMIA is available through a limited program under a Risk Evaluation and Mitigation Strategy (REMS) ( 5.1).
- Increase in Heart Rate: Monitor heart rate, especially in those with cardiac or cerebrovascular disease ( 5.2).
- Suicidal Behavior and Ideation: Monitor for depression or suicidal thoughts. Discontinue QSYMIA if symptoms develop ( 5.3).
- Risk of Ophthalmologic Adverse Reactions: Acute myopia and secondary angle closure glaucoma have been reported. Immediately discontinue QSYMIA if symptoms develop. Consider QSYMIA discontinuation if visual field defects occur ( 5.4).
- Mood and Sleep Disorders: Consider dosage reduction or discontinuation for clinically significant or persistent mood or sleep disorder symptoms ( 5.5).
- Cognitive Impairment: May cause disturbances in attention or memory, or speech/language problems. Caution patients about operating automobiles or hazardous machinery when starting treatment ( 5.6).
- Slowing of Linear Growth: Consider dosage reduction or discontinuation if pediatric patients are not growing or gaining height as expected ( 5.7).
- Metabolic Acidosis: Measure electrolytes before and during treatment. If persistent metabolic acidosis develops, reduce dosage or discontinue QSYMIA ( 5.8).
- Decrease in Renal Function: Measure creatinine before and during treatment. For persistent creatinine elevations, reduce dosage or discontinue QSYMIA ( 5.9).
- Serious Skin Reactions:QSYMIA should be discontinued at the first sign of a rash, unless the rash is clearly not drug-related ( 5.16).
Adverse Reactions/Side Effects
Most common adverse reactions in:
- Adults (incidence ≥ 5% and at least 1.5 times placebo) are: paraesthesia, dizziness, dysgeusia, insomnia, constipation, and dry mouth
- Pediatric patients aged 12 years and older (incidence ≥4% and greater than placebo) are: depression, dizziness, arthralgia, pyrexia, influenza, and ligament sprain ( 6.1).
To report SUSPECTED ADVERSE REACTIONS, contact VIVUS LLC, at 1-888-998-4887 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Oral Contraceptives: Altered exposure of progestin and estrogen may cause irregular bleeding, but not increased risk of pregnancy. Advise patients not to discontinue oral contraceptives if spotting occurs ( 7).
- CNS Depressants Including Alcohol: May potentiate CNS depressant effects. Avoid excessive use of alcohol ( 7).
- Non-potassium Sparing Diuretics: May potentiate hypokalemia. Measure potassium before and during treatment ( 7).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2023
Related/similar drugs
Mounjaro, semaglutide, phentermine, Wegovy, Saxenda, Alli
Full Prescribing Information
1. Indications and Usage for Qsymia
QSYMIA is indicated as an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in:
- Adults with an initial body mass index (BMI) of:
- 30 kg/m 2or greater (obese), or
- 27 kg/m 2or greater (overweight) in the presence of at least one weight-related comorbidity such as hypertension, type 2 diabetes mellitus, or dyslipidemia
- Pediatric patients aged 12 years and older with an initial BMI in the 95 thpercentile or greater standardized for age and sex.
Limitations of Use
- The effect of QSYMIA on cardiovascular morbidity and mortality has not been established.
- The safety and effectiveness of QSYMIA in combination with other products intended for weight loss, including prescription drugs, over-the-counter drugs, and herbal preparations, have not been established.
2. Qsymia Dosage and Administration
2.1 Patient Selection
Adults
Select adult patients for QSYMIA treatment based on BMI [see Indications and Usage (1)] . Determine the patient's BMI by dividing weight (in kilograms) by height (in meters) squared. A BMI conversion table based on height [inches (in) or centimeters (cm)] and weight [pounds (lb) or kilograms (kg)] is provided below (see Table 1).
| Table 1. BMI Conversion Chart |
|---|
 |
Pediatric Patients Aged 12 Years and Older
Select pediatric patients for QSYMIA treatment based on BMI percentile [see Indications and Usage (1)] . See Table 2for BMI percentiles by age and sex for pediatric patients aged 12 years and older.
| Male | Female | |
|---|---|---|
| Age (in years) | 95th Percentile BMI Value | 95th Percentile BMI Value |
| 12 | 24.2 | 25.3 |
| 12.5 | 24.7 | 25.8 |
| 13 | 25.2 | 26.3 |
| 13.5 | 25.6 | 26.8 |
| 14 | 26.0 | 27.3 |
| 14.5 | 26.5 | 27.7 |
| 15 | 26.8 | 28.1 |
| 15.5 | 27.2 | 28.5 |
| 16 | 27.6 | 28.9 |
| 16.5 | 27.9 | 29.3 |
| 17 | 28.3 | 29.6 |
| 17.5 | 28.6 | 30.0 |
2.2 Recommended Testing Prior to and During Treatment with QSYMIA
Prior to QSYMIA initiation and during treatment with QSYMIA, the following is recommended:
- Obtain a negative pregnancy test before initiating QSYMIA in patients who can become pregnant and monthly during QSYMIA therapy. QSYMIA is contraindicated during pregnancy [see Contraindications (4), Warnings and Precautions (5.1)and Use in Specific Populations (8.3)].
- Obtain a blood chemistry profile that includes bicarbonate, creatinine, and potassium in all patients, and glucose in patients with type 2 diabetes on antidiabetic medication prior to initiating QSYMIA treatment and periodically during treatment [Warnings and Precautions (5.8, 5.9, 5.10, 5.15)].
2.3 Recommended Dosage and Administration
The recommended dosage, titration, and administration of QSYMIA are as follows:
- Take QSYMIA orally once daily in the morning with or without food. Avoid administration of QSYMIA in the evening due to the possibility of insomnia.
- The recommended starting dosage is QSYMIA 3.75 mg/23 mg (phentermine 3.75 mg/topiramate 23 mg) orally once daily for 14 days; after 14 days increase to the recommended dosage of QSYMIA 7.5 mg/46 mg (phentermine 7.5 mg/topiramate 46 mg) orally once daily.
- After 12 weeks of treatment with QSYMIA 7.5 mg/46 mg, evaluate weight loss for adults or BMI reduction for pediatric patients aged 12 years and older. If an adult patient has not lost at least 3% of baseline body weight or a pediatric patient has not experienced a reduction of at least 3% of baseline BMI, increase the dosage to QSYMIA 11.25 mg/69 mg (phentermine 11.25 mg/topiramate 69 mg) orally once daily for 14 days; followed by an increase in the dosage to QSYMIA 15 mg/92 mg (phentermine 15 mg/topiramate 92 mg) orally once daily.
- After 12 weeks of treatment with QSYMIA 15 mg/92 mg, evaluate weight loss for adults or BMI reduction for pediatric patients aged 12 years and older. If an adult patient has not lost at least 5% of baseline body weight or a pediatric patient has not experienced a reduction of at least 5% of baseline BMI, discontinue QSYMIA [see Dosage and Administration (2.4)] , as it is unlikely that the patient will achieve and sustain clinically meaningful weight loss with continued treatment.
- Monitor the rate of weight loss in pediatric patients. If weight loss exceeds 2 lbs (0.9 kg)/week, consider dosage reduction.
2.4 Discontinuation of QSYMIA 15 mg/92 mg
Discontinue QSYMIA 15 mg/92 mg gradually by taking QSYMIA 15 mg/92 mg once daily every other day for at least 1 week prior to stopping treatment altogether, due to the possibility of precipitating a seizure [see Warnings and Precautions (5.12)and Drug Abuse and Dependence (9.3)].
2.5 Recommended Dosage in Patients with Renal Impairment
Avoid use of QSYMIA in patients with end-stage renal disease on dialysis.
In patients with severe (creatinine clearance [CrCl] less than 30 mL/min) or moderate (CrCl greater than or equal to 30 and less than 50 mL/min) renal impairment (CrCl calculated using the Cockcroft-Gault equation with actual body weight), the maximum recommended dosage is QSYMIA 7.5 mg/46 mg once daily.
The recommended dosage in patients with mild (CrCl greater or equal to 50 and less than 80 mL/min) renal impairment is the same as the recommended dosage for patients with normal renal function [see Warnings and Precautions (5.9), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)] .
2.6 Recommended Dosage in Patients with Hepatic Impairment
Avoid use of QSYMIA in patients with severe hepatic impairment (Child-Pugh score 10 - 15). In patients with moderate hepatic impairment (Child-Pugh score 7 - 9), the maximum recommended dosage is QSYMIA 7.5 mg/46 mg once daily.
The recommended dosage of QSYMIA in patients with mild hepatic impairment (Child-Pugh 5 - 6) is the same as the recommended dosage in patients with normal hepatic function [see Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)] .
3. Dosage Forms and Strengths
QSYMIA extended-release capsules are available in four strengths (phentermine mg/topiramate mg):
- 3.75 mg/23 mg - purple cap imprinted with VIVUS and purple body imprinted with 3.75/23
- 7.5 mg/46 mg - purple cap imprinted with VIVUS and yellow body imprinted with 7.5/46
- 11.25 mg/69 mg - yellow cap imprinted with VIVUS and yellow body imprinted with 11.25/69
- 15 mg/92 mg - yellow cap imprinted with VIVUS and white body imprinted with 15/92
4. Contraindications
QSYMIA is contraindicated in patients:
- Who are pregnant [see Warnings and Precautions (5.1)and Use in Specific Populations (8.1)]
- With glaucoma [see Warnings and Precautions (5.4)]
- With hyperthyroidism
- Taking or within 14 days of stopping a monoamine oxidase inhibitors [see Drug Interactions (7)]
- With known hypersensitivity to phentermine, topiramate or excipient in QSYMIA, or idiosyncrasy to the sympathomimetic amines [see Adverse Reactions (6.2)].
5. Warnings and Precautions
5.1 Embryo-Fetal Toxicity
QSYMIA can cause fetal harm. Data from pregnancy registries and epidemiologic studies indicate that a fetus exposed to topiramate in the first trimester of pregnancy has an increased risk of major congenital malformations, including but not limited to cleft lip and/or cleft palate (oral clefts), and of being small for gestational age (SGA). When multiple species of pregnant animals received topiramate at clinically relevant doses, structural malformations, including craniofacial defects, and reduced fetal weights occurred in offspring. A negative pregnancy test is recommended before initiating QSYMIA treatment in patients who can become pregnant and monthly during QSYMIA therapy. Advise patients who can become pregnant of the potential risk to a fetus and to use effective contraception during QSYMIA therapy [see Use in Specific Populations (8.1, 8.3)] .
QSYMIA Risk Evaluation and Mitigation Strategy (REMS)
Because of the teratogenic risk associated with QSYMIA therapy, QSYMIA is available through a limited program under the REMS. Under the QSYMIA REMS, only certified pharmacies may distribute QSYMIA. Further information is available at www.QSYMIAREMS.com or by telephone at 1-888-998-4887.
5.2 Increase in Heart Rate
QSYMIA can cause an increase in resting heart rate. A higher percentage of QSYMIA-treated adults and pediatric patients aged 12 years and older experienced heart rate increases from baseline of more than 5, 10, 15, and 20 beats per minute (bpm) compared to placebo-treated patients [see Adverse Reactions (6.1)] . The clinical significance of a heart rate elevation with QSYMIA treatment is unclear, especially for patients with cardiac and cerebrovascular disease.
Measure resting heart rate regularly in all patients taking QSYMIA, especially patients with cardiac or cerebrovascular disease and when initiating or increasing the dosage of QSYMIA. QSYMIA has not been studied in patients with recent or unstable cardiac or cerebrovascular disease and therefore use is not recommended.
Advise patients to inform their healthcare provider of palpitations or feelings of a racing heartbeat while at rest during QSYMIA treatment. For patients who experience a sustained increase in resting heart rate while taking QSYMIA, reduce the dosage or discontinue QSYMIA [see Warnings and Precautions (5.12)] .
5.3 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including topiramate, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Pooled analyses of 199 placebo-controlled clinical studies (monotherapy and adjunctive therapy, median treatment duration 12 weeks) of 11 different AEDs across several indications showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI 1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. The estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in AED-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about AED effect on suicide. The increased risk of suicidal thoughts or behavior with AEDs was observed as early as 1 week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed. The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age in the clinical trials analyzed.
In a QSYMIA clinical trial of pediatric patients aged 12 years and older, 1 (0.6%) of the 167 QSYMIA-treated patients reported suicidal ideation and behavior which required hospitalization. No placebo-treated patients reported suicidal behavior or ideation.
Monitor all patients for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior. Discontinue QSYMIA in patients who experience suicidal thoughts or behaviors [see Warnings and Precautions (5.12)] . Avoid QSYMIA in patients with a history of suicidal attempts or active suicidal ideation.
5.4 Risk of Ophthalmologic Adverse Reactions
Acute Myopia and Secondary Angle Closure Glaucoma
A syndrome consisting of acute myopia associated with secondary angle closure glaucoma has been reported in patients treated with topiramate. Symptoms include acute onset of decreased visual acuity and/or ocular pain. Ophthalmologic findings can include myopia, mydriasis, anterior chamber shallowing, ocular hyperemia (redness), choroidal detachments, retinal pigment epithelial detachments, macular striae, and increased intraocular pressure. This syndrome may be associated with supraciliary effusion resulting in anterior displacement of the lens and iris, with secondary angle closure glaucoma. Symptoms typically occur within 1 month of initiating treatment with topiramate but may occur at any time during therapy. In contrast to primary narrow angle glaucoma, which is rare under 40 years of age, secondary angle closure glaucoma associated with topiramate has been reported in pediatric patients as well as adults. The primary treatment to reverse symptoms is discontinuation of QSYMIA as rapidly as possible in consultation with the treating physician. Elevated intraocular pressure of any etiology, if left untreated, can lead to serious sequelae including permanent loss of vision.
Visual Field Defects
Visual field defects (independent of elevated intraocular pressure) have been reported in clinical trials and in postmarketing experience in patients receiving topiramate. In clinical trials, most of these events were reversible after topiramate discontinuation. If visual problems occur at any time during treatment, consider discontinuing QSYMIA.
5.5 Mood and Sleep Disorders
QSYMIA can cause mood disorders, including depression and anxiety, as well as insomnia. Patients with a history of depression may be at increased risk of recurrent depression or other mood disorders while taking QSYMIA [see Adverse Reactions (6.1)].
Consider dosage reduction or discontinuation of QSYMIA if clinically significant or persistent symptoms occur. Discontinue QSYMIA if patients have symptoms of suicidal ideation or behavior [see Warnings and Precautions (5.3)] .
5.6 Cognitive Impairment
QSYMIA can cause cognitive dysfunction (e.g., impairment of concentration/attention, difficulty with memory, and speech or language problems, particularly word-finding difficulties). Rapid titration or high initial doses of QSYMIA may be associated with higher rates of cognitive events such as attention, memory, and language/word-finding difficulties [see Adverse Reactions (6.1)] . The concomitant use of alcohol or central nervous system (CNS) depressant drugs with QSYMIA may potentiate CNS depression or other centrally mediated effects of these agents, such as dizziness, cognitive adverse reactions, drowsiness, light-headedness, impaired coordination, and somnolence.
Caution patients about operating hazardous machinery, including automobiles, until they are reasonably certain QSYMIA therapy does not affect them adversely. Caution patients against excessive alcohol intake while receiving QSYMIA.
If cognitive dysfunction persists, consider dosage reduction or discontinuation of QSYMIA [see Warnings and Precautions (5.12)] .
5.7 Slowing of Linear Growth
QSYMIA is associated with a reduction in height velocity (centimeters of height gained per year) in obese pediatric patients 12 to 17 years of age. In a 56-week study, average height increased from baseline in both QSYMIA- and placebo-treated patients; however, a lower height velocity of -1.3 to -1.4 cm/year was observed in QSYMIA-treated compared to placebo-treated patients. Monitor height velocity in pediatric patients treated with QSYMIA. Consider dosage reduction or discontinuation of QSYMIA if pediatric patients are not growing or gaining height as expected [see Warnings and Precautions (5.12)] .
5.8 Metabolic Acidosis
Hyperchloremic, non-anion gap, metabolic acidosis (decreased serum bicarbonate below the normal reference range in the absence of chronic respiratory alkalosis) has been reported in patients treated with QSYMIA [see Adverse Reactions (6.1)] . Manifestations of acute or chronic metabolic acidosis may include hyperventilation, nonspecific symptoms such as fatigue and anorexia, or more severe sequelae including cardiac arrhythmias or stupor. Chronic, untreated metabolic acidosis may increase the risk for nephrolithiasis or nephrocalcinosis and may also result in osteomalacia (referred to as rickets in pediatric patients) and/or osteoporosis with an increased risk for fractures. Chronic metabolic acidosis in pediatric patients may also reduce growth rates, which may decrease the maximal height achieved.
Conditions or therapies that predispose to acidosis (i.e., renal disease, severe respiratory disorders, status epilepticus, diarrhea, surgery, or ketogenic diet) may be additive to the bicarbonate lowering effects of QSYMIA. Concomitant use of QSYMIA and a carbonic anhydrase inhibitor may increase the severity of metabolic acidosis and may also increase the risk of kidney stone formation [see Warnings and Precautions (5.13)] . Avoid use of QSYMIA with other carbonic anhydrase inhibitors. If concomitant use of QSYMIA with another carbonic anhydrase inhibitor is unavoidable, the patient should be monitored for the appearance or worsening of metabolic acidosis.
Measure electrolytes including serum bicarbonate prior to starting QSYMIA and during QSYMIA treatment. In QSYMIA clinical trials, the peak reduction in serum bicarbonate typically occurred within 4 weeks of titration to the assigned dose, and in most patients there was a correction of bicarbonate by week 56, without any dosage reduction. However, if persistent metabolic acidosis develops while taking QSYMIA, reduce the dosage or discontinue QSYMIA [see Warnings and Precautions (5.12)] .
5.9 Decrease in Renal Function
QSYMIA can cause an increase in serum creatinine that reflects a decrease in renal function (glomerular filtration rate). In clinical trials, peak increases in serum creatinine were observed after 4 to 8 weeks of treatment. On average, serum creatinine gradually declined but remained elevated over baseline creatinine values. The changes in serum creatinine (and measured GFR) with short-term (4-weeks) QSYMIA treatment appear reversible with treatment discontinuation, but the effect of chronic treatment on renal function is not known.
Measure serum creatinine prior to starting QSYMIA and during QSYMIA treatment. If persistent elevations in creatinine occur, reduce the dosage or discontinue QSYMIA [see Warnings and Precautions (5.12), Adverse Reactions (6.1), Clinical Pharmacology (12.2)].
5.10 Risk of Hypoglycemia in Patients with Type 2 Diabetes Mellitus on Antidiabetic Therapy
Weight loss may increase the risk of hypoglycemia in patients with type 2 diabetes mellitus treated with insulin and/or insulin secretagogues (e.g., sulfonylureas). QSYMIA has not been studied in combination with insulin. Measure blood glucose levels prior to starting QSYMIA and during QSYMIA treatment in patients with type 2 diabetes on antidiabetic medication. The risk of hypoglycemia may be lowered by a reduction of the dosage of insulin and/or insulin secretagogues. If a patient develops hypoglycemia after starting QSYMIA, appropriate changes should be made to the antidiabetic drug regimen.
5.11 Risk of Hypotension in Patients Treated with Antihypertensive Medications
In hypertensive patients being treated with antihypertensive medications, weight loss may increase the risk of hypotension and associated symptoms including dizziness, lightheadedness, and syncope. Measure blood pressure prior to starting QSYMIA and during QSYMIA treatment in patients being treated for hypertension. If a patient develops symptoms associated with low blood pressure after starting QSYMIA, appropriate changes should be made to the antihypertensive drug regimen.
5.12 Risk of Seizures with Abrupt Withdrawal of QSYMIA
Abrupt withdrawal of topiramate has been associated with seizures in individuals without a history of seizures or epilepsy. In situations where immediate termination of QSYMIA is medically required, appropriate monitoring is recommended. Patients discontinuing QSYMIA 15 mg/92 mg should be gradually tapered to reduce the possibility of precipitating a seizure [see Dosage and Administration (2.4)and Drug Abuse and Dependence (9.3)] .
5.13 Kidney Stones
QSYMIA has been associated with kidney stone formation [see Adverse Reactions (6.1)] . Topiramate inhibits carbonic anhydrase activity and promotes kidney stone formation by reducing urinary citrate excretion and increasing urine pH. Patients on a ketogenic diet may be at increased risk for kidney stone formation. An increase in urinary calcium and a marked decrease in urinary citrate was observed in topiramate-treated pediatric patients in a one-year, active-controlled study. Increased ratio of urinary calcium/citrate increases the risk of kidney stones and/or nephrocalcinosis.
Avoid the use of QSYMIA with other drugs that inhibit carbonic anhydrase [see Drug Interactions (7)] . Advise patients to increase fluid intake (to increase urinary output), which may decrease the concentration of substances involved in kidney stone formation .
5.14 Oligohidrosis and Hyperthermia
Oligohidrosis (decreased sweating), infrequently resulting in hospitalization, has been reported in association with the use of topiramate. Decreased sweating and an elevation in body temperature above normal characterized these cases. Some of the cases have been reported with topiramate after exposure to elevated environmental temperatures.
The majority of the reports associated with topiramate have been in pediatric patients. Advise all patients and caregivers to monitor for decreased sweating and increased body temperature during physical activity, especially in hot weather. Patients on concomitant medications that predispose them to heat-related disorders may be at increased risk.
5.15 Hypokalemia
QSYMIA can increase the risk of hypokalemia through its inhibition of carbonic anhydrase activity. In addition, when QSYMIA is used in conjunction with non-potassium sparing diuretics this may further potentiate potassium-wasting. Measure potassium before and during treatment with QSYMIA. [see Adverse Reactions (6.1), Drug Interactions (7), and Clinical Pharmacology (12.3)] .
5.16 Serious Skin Reactions
Serious skin reactions (Stevens-Johnson Syndrome [SJS] and Toxic Epidermal Necrolysis [TEN]) have been reported in patients receiving topiramate. QSYMIA should be discontinued at the first sign of a rash, unless the rash is clearly not drug-related. If signs or symptoms suggest SJS/TEN, use of this drug should not be resumed and alternative therapy should be considered. Inform patients about the signs of serious skin reactions.
5.17 Allergic Reactions Due to Inactive Ingredient FD&C Yellow No. 5
This product contains FD&C Yellow No. 5 (tartrazine) which may cause allergic-type reactions (including bronchial asthma) in certain susceptible persons. Although the overall incidence of FD&C Yellow No. 5 (tartrazine) sensitivity in the general population is low, it is frequently seen in patients who also have aspirin hypersensitivity.
6. Adverse Reactions/Side Effects
The following important adverse reactions are described elsewhere in the labeling:
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.1)and Use in Specific Populations (8.1, 8.6)]
- Increase in Heart Rate [ see Warnings and Precautions (5.2)]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.3)]
- Risk of Ophthalmologic Adverse Reactions [see Warnings and Precautions (5.4)]
- Mood and Sleep Disorders [see Warnings and Precautions (5.5)]
- Cognitive Impairment [see Warnings and Precautions (5.6)]
- Slowing of Linear Growth [see Warnings and Precautions (5.7)]
- Metabolic Acidosis [see Warnings and Precautions (5.8)]
- Decrease in Renal Function [see Warnings and Precautions (5.9)]
- Risk of Seizures with Abrupt Withdrawal of QSYMIA [see Warnings and Precautions (5.12)]
- Kidney Stones [see Warnings and Precautions (5.13)]
- Oligohydrosis and Hyperthermia [see Warnings and Precautions (5.14)]
- Hypokalemia [see Warnings and Precautions (5.15)]
- Serious Skin Reactions [see Warnings and Precautions (5.16)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The data described herein reflects exposure to QSYMIA in two 1-year, randomized, double-blind, placebo-controlled, multicenter clinical trials and two supportive trials in 2318 adult patients with overweight or obesity (936 [40%] patients with hypertension, 309 [13%] patients with type 2 diabetes, 808 [35%] patients with BMI greater than 40 kg/m 2) exposed for a mean duration of 298 days. Data in this section also describe adverse reactions from a 1-year, randomized, double-blind, placebo-controlled multicenter clinical trial that evaluated 223 pediatric patients (12 to 17 years old) with obesity [see Clinical Studies (14)] .
Adults
Adverse reactions occurring at greater than or equal to 5% and at least 1.5 times placebo in adults include paraesthesia, dizziness, dysgeusia, insomnia, constipation, and dry mouth.
Adverse reactions reported in greater than or equal to 2% of QSYMIA-treated adults and more frequently than in the placebo group are shown in Table 3.
| Preferred Term | Placebo
(N = 1561) % | QSYMIA
3.75 mg/23 mg (N = 240) % | QSYMIA
7.5 mg/46 mg (N = 498) % | QSYMIA
15 mg/92 mg (N = 1580) % |
|---|---|---|---|---|
| Paraesthesia | 2 | 4 | 14 | 20 |
| Dry Mouth | 3 | 7 | 14 | 19 |
| Constipation | 6 | 8 | 15 | 16 |
| Upper Respiratory Tract Infection | 13 | 16 | 12 | 14 |
| Headache | 9 | 10 | 7 | 11 |
| Dysgeusia | 1 | 1 | 7 | 9 |
| Insomnia | 5 | 5 | 6 | 9 |
| Nasopharyngitis | 8 | 13 | 11 | 9 |
| Dizziness | 3 | 3 | 7 | 9 |
| Sinusitis | 6 | 8 | 7 | 8 |
| Nausea | 4 | 6 | 4 | 7 |
| Back Pain | 5 | 5 | 6 | 7 |
| Fatigue | 4 | 5 | 4 | 6 |
| Diarrhea | 5 | 5 | 6 | 6 |
| Vision Blurred | 4 | 6 | 4 | 5 |
| Bronchitis | 4 | 7 | 4 | 5 |
| Urinary Tract Infection | 4 | 3 | 5 | 5 |
| Cough | 4 | 3 | 4 | 5 |
| Influenza | 4 | 8 | 5 | 4 |
| Depression | 2 | 3 | 3 | 4 |
| Anxiety | 2 | 3 | 2 | 4 |
| Hypoesthesia | 1 | 1 | 4 | 4 |
| Irritability | 1 | 2 | 3 | 4 |
| Alopecia | 1 | 2 | 3 | 4 |
| Disturbance in Attention | 1 | 0 | 2 | 4 |
| Pain in Extremity | 3 | 2 | 3 | 3 |
| Muscle Spasms | 2 | 3 | 3 | 3 |
| Dyspepsia | 2 | 2 | 2 | 3 |
| Gastroesophageal Reflux Disease | 1 | 1 | 3 | 3 |
| Rash | 2 | 2 | 2 | 3 |
| Hypokalemia | 0 | 0 | 1 | 3 |
| Dry Eye | 1 | 1 | 1 | 3 |
| Gastroenteritis | 2 | 1 | 2 | 3 |
| Pharyngolaryngeal Pain | 2 | 3 | 1 | 2 |
| Paraesthesia Oral | 0 | 0 | 1 | 2 |
| Eye Pain | 1 | 2 | 2 | 2 |
| Nasal Congestion | 1 | 2 | 1 | 2 |
| Thirst | 1 | 2 | 2 | 2 |
| Sinus Congestion | 2 | 3 | 3 | 2 |
| Procedural Pain | 2 | 2 | 2 | 2 |
| Palpitations | 1 | 1 | 2 | 2 |
| Musculoskeletal Pain | 1 | 1 | 3 | 2 |
| Decreased Appetite | 1 | 2 | 2 | 2 |
| Neck Pain | 1 | 1 | 2 | 1 |
| Dysmenorrhea | 0 | 2 | 0 | 1 |
| Chest Discomfort | 0 | 2 | 0 | 1 |
Pediatric Patients Aged 12 Years and Older
Adverse reactions occurring in pediatric patients treated with either QSYMIA 15 mg/92 mg or QSYMIA 7.5 mg/46 mg at greater than or equal to 4% and higher than placebo include depression, pyrexia, dizziness, arthralgia, influenza, and ligament sprain.
Adverse reactions reported in greater than or equal to 2% of QSYMIA-treated pediatric patients and more frequently than in the placebo group from a study in pediatric patients aged 12 years and older are shown in Table 4.
| Preferred Term | Placebo
(N = 56) % | QSYMIA
7.5 mg/46 mg (N = 54) % | QSYMIA
15 mg/92 mg (N = 113) % |
|---|---|---|---|
| Depression | 0 | 2 | 4 |
| Nausea | 4 | 4 | 4 |
| Pyrexia | 2 | 2 | 4 |
| Dizziness | 0 | 2 | 4 |
| Arthralgia | 0 | 2 | 4 |
| Paraesthesia | 0 | 2 | 3 |
| Anxiety | 0 | 2 | 3 |
| Abdominal Pain Upper | 0 | 0 | 3 |
| Fatigue | 2 | 0 | 3 |
| Ear Infection | 0 | 2 | 3 |
| Musculoskeletal Chest Pain | 0 | 0 | 3 |
| Influenza | 0 | 4 | 2 |
| Ligament Sprain | 0 | 4 | 2 |
Increase in Heart Rate
In adult and pediatric clinical trials, there was a higher incidence of heart rate elevations observed in QSYMIA-treated compared to placebo-treated patients.
Table 5 and Table 6 provide the numbers and percentages of adult and pediatric patients, respectively, with elevations in heart rate in clinical studies of up to one year.
| Placebo
N=1561 n (%) | QSYMIA
3.75 mg/23 mg N=240 n (%) | QSYMIA
7.5 mg/46 mg N=498 n (%) | QSYMIA
15 mg/92 mg N=1580 n (%) |
|
|---|---|---|---|---|
| Greater than 5 bpm | 1021 (65.4) | 168 (70.0) | 372 (74.7) | 1228 (77.7) |
| Greater than 10 bpm | 657 (42.1) | 120 (50.0) | 251 (50.4) | 887 (56.1) |
| Greater than 15 bpm | 410 (26.3) | 79 (32.9) | 165 (33.1) | 590 (37.3) |
| Greater than 20 bpm | 186 (11.9) | 36 (15.0) | 67 (13.5) | 309 (19.6) |
| Placebo
N=56 n (%) | QSYMIA
7.5 mg/46 mg N=54 n (%) | QSYMIA
15 mg/92 mg N=113 n (%) |
|
|---|---|---|---|
| Greater than 5 bpm | 37 (66.1) | 38 (70.4) | 92 (81.4) |
| Greater than 10 bpm | 26 (46.4) | 30 (55.6) | 73 (64.6) |
| Greater than 15 bpm | 17 (30.4) | 18 (33.3) | 48 (42.5) |
| Greater than 20 bpm | 10 (17.9) | 10 (18.5) | 27 (23.9) |
Paraesthesia/Dysgeusia
In adult clinical trials, reports of paraesthesia, characterized as tingling in hands, feet, or face, and dysgeusia, characterized as a metallic taste, occurred (see Table 3). Adverse reactions of paraesthesia were also reported in pediatric patients (see Table 4). QSYMIA-treated adult patients discontinued treatment due to these adverse reactions (1% for paraesthesia and 0.6% for dysgeusia); no pediatric patients discontinued treatment due to paraesthesia or dysgeusia.
Mood and Sleep Disorders
The proportion of adult patients in 1-year controlled trials of QSYMIA reporting one or more adverse reactions related to mood and sleep disorders was 15% and 21% with QSYMIA 7.5 mg/46 mg and 15 mg/92 mg, respectively, compared to 10% with placebo. These events were further categorized into sleep disorders, anxiety, and depression. Reports of sleep disorders were typically characterized as insomnia and occurred in 8.1% and 11% of patients treated with QSYMIA 7.5 mg/46 mg and 15 mg/92 mg, respectively, compared to 5.8% of patients treated with placebo. Reports of anxiety occurred in 4.8% and 7.9% of patients treated with QSYMIA 7.5 mg/46 mg and 15 mg/92 mg, respectively, compared to 2.6% of patients treated with placebo. Reports of depression/mood problems occurred in 3.8% and 7.6% of patients treated with QSYMIA 7.5 mg/46 mg and 15 mg/92 mg, respectively, compared to 3.4% of patients treated with placebo. Mood and sleep disorder adverse reactions occurred in patients with and without a history of depression.
In a pediatric clinical trial, higher proportions of QSYMIA-treated patients reported one or more adverse reactions related to mood (e.g., depression, anxiety) and sleep disorders (e.g., insomnia) compared to placebo-treated patients (See Table 4).
Cognitive Disorders
In the 1-year controlled trials of QSYMIA in adults, the proportion of patients who experienced one or more cognitive-related adverse reactions was 5.0% for QSYMIA 7.5 mg/46 mg and 7.6% for QSYMIA 15 mg/92 mg, compared to 1.5% for placebo. These adverse reactions were comprised primarily of reports of problems with attention/concentration, memory, and language (word-finding). These events occurred at any time during treatment with QSYMIA.
Slowing of Linear Growth
QSYMIA is associated with a reduction in height velocity (centimeters of height gained per year) in obese pediatric patients 12 to 17 years of age. In a 56-week study, average height increased from baseline in both QSYMIA- and placebo-treated patients; however, a lower height velocity of -1.3 to -1.4 cm/year was observed in QSYMIA-treated compared to placebo-treated patients
Decrease in Bone Mineral Density
QSYMIA is associated with less bone mineral acquisition in pediatric patients 12 to 17 years of age. In a substudy (n=66) evaluating bone mineralization via dual-energy X-ray absorptiometry (DEXA), increases in bone mineral density (BMD) at the lumbar spine and total body less head (TBLH) were smaller in pediatric patients treated with QSYMIA compared to those treated with placebo after 1 year of treatment. Declines in BMD Z-scores of -0.5 or greater from baseline for TBLH were observed in 9% of QSYMIA 7.5 mg/46 mg-treated patients and 30% of QSYMIA 15 mg/92 mg-treated patients, compared to 0% of placebo-treated patients. The sample size and study duration were too small to determine if fracture risk is increased. Decreased BMD was not correlated with decreased serum bicarbonate, which commonly occurs with QSYMIA treatment, or changes in body weight. No patient had a BMD Z-score that went below -2.0 during the trial. Similar findings were observed in a 1 year, active-controlled trial of topiramate in pediatric patients with another condition.
Nephrolithiasis
In the 1-year controlled trials of QSYMIA in adults, the incidence of nephrolithiasis was 0.2% for QSYMIA 7.5 mg/46 mg and 1.2% for QSYMIA 15 mg/92 mg, compared to 0.3% for placebo.
Laboratory Abnormalities
Serum Bicarbonate
In the 1-year controlled trials of QSYMIA in adults, the incidence of persistent decreases in serum bicarbonate below the normal range (levels of less than 21 mEq/L at 2 consecutive visits or at the final visit) was 6.4% for QSYMIA 7.5 mg/46 mg and 12.8% for QSYMIA 15 mg/92 mg, compared to 2.1% for placebo. The incidence of persistent, markedly low serum bicarbonate values (levels of less than 17 mEq/L on 2 consecutive visits or at the final visit) was 0.2% for QSYMIA 7.5 mg/46 mg dose and 0.7% for QSYMIA 15 mg/92 mg dose, compared to 0.1% for placebo. In a pediatric clinical trial, 60 to 70% QSYMIA-treated patients had a persistent bicarbonate level below the normal range (<21 mEq/L) compared to 43% of placebo-treated patients.
Serum Potassium
In the 1-year controlled trials of QSYMIA in adults, the incidence of persistent low serum potassium values (less than 3.5 mEq/L at two consecutive visits or at the final visit) during the trial was 3.6% for QSYMIA 7.5 mg/46 mg dose and 4.9% for QSYMIA 15 mg/92 mg, compared to 1.1% for placebo. Of the subjects who experienced persistent low serum potassium, 88% were receiving treatment with a non-potassium sparing diuretic.
The incidence of markedly low serum potassium (less than 3 mEq/L, and a reduction from pre-treatment of greater than 0.5 mEq/L) at any time during the trial was 0.2% for QSYMIA 7.5 mg/46 mg dose and 0.7% for QSYMIA 15 mg/92 mg dose, compared to 0.0% for placebo. Persistent markedly low serum potassium (less than 3 mEq/L, and a reduction from pre-treatment of greater than 0.5 mEq/L at two consecutive visits or at the final visit) occurred in 0.2% receiving QSYMIA 7.5 mg/46 mg dose and 0.1% receiving QSYMIA 15 mg/92 mg dose, compared to 0.0% receiving placebo.
Low serum potassium levels (<3.5 mEq/L) were not observed in a 56-week clinical trial of pediatric patients with obesity.
Serum Creatinine
In the 1-year controlled trials of QSYMIA in adults and pediatric patients, there was an increase in serum creatinine from baseline, peaking between Week 4 to 8 in adults and at Week 16 in pediatric patients. Serum creatinine values declined but remained elevated over baseline over 1 year of treatment. The incidence of increases in serum creatinine of greater than or equal to 0.3 mg/dL at any time during treatment in adults was 7.2% for QSYMIA 7.5 mg/46 mg and 8.4% for QSYMIA 15 mg/92 mg, compared to 2.0% for placebo; 17% of pediatric patients treated with QSYMIA 7.5 mg/46 mg or QSYMIA 15 mg/92 mg and 0% of patients treated with placebo had a serum creatinine ≥0.3 mg/dL at any time post-randomization. Increases in serum creatinine of greater than or equal to 50% over baseline occurred in 2.0% of adult subjects receiving QSYMIA 7.5 mg/46 mg and 2.8% receiving QSYMIA 15 mg/92 mg, compared to 0.6% receiving placebo.
Serum Ammonia
Hyperammonemia with or without encephalopathy has been reported with topiramate. The risk for hyperammonemia with topiramate appears dose related and has been reported more frequently when concomitantly used with valproic acid [see Drug Interactions (7)].
The incidence of hyperammonemia in pediatric patients 12 to 17 years of age in clinical trials of another condition was 26% in patients taking topiramate at 100 mg/day (1.1 times the maximum recommended dosage of QSYMIA) and 14% in patients taking topiramate at 50 mg/day (0.6 times the maximum recommended dosage of QSYMIA), compared to 9% in patients taking placebo. There was also an increased incidence of markedly increased hyperammonemia (defined as 50% above the upper limit of normal reference range) at the 100 mg dose.
6.2 Postmarketing Experience
The following adverse reactions have been reported during post approval use of QSYMIA, phentermine, and topiramate. Because these reactions are reported voluntarily from a population of uncertain size it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
QSYMIA
Psychiatric:suicidal ideation, suicidal behavior
Ophthalmic:acute angle closure glaucoma, increased intraocular pressure
Phentermine
Allergic Reactions:urticaria
Cardiovascular:elevation of blood pressure, ischemic events
Central Nervous System:euphoria, psychosis, tremor
Reproductive:changes in libido, impotence
Topiramate
Dermatologic:bullous skin reactions (including erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis), pemphigus
Gastrointestinal:pancreatitis
Hepatic:hepatic failure (including fatalities), hepatitis
Metabolic:hyperammonemia with or without encephalopathy has been reported with concomitant valproic acid [see Drug Interactions (7)], hypothermia
Ophthalmic:maculopathy
7. Drug Interactions
Table 7 displays clinically significant drug interactions with QSYMIA.
| Monamine Oxidase Inhibitors | |
| Clinical Impact | Concomitant use of phentermine with monoamine oxidase inhibitors (MAOIs) increases the risk of hypertensive crisis. |
| Intervention | Concomitant use of QSYMIA is contraindicated during MAOI treatment and within 14 days of stopping an MAOI. |
| Oral Contraceptives | |
| Clinical Impact | Co-administration of multiple-dose QSYMIA 15 mg/92 mg once daily with a single dose of oral contraceptive containing 35 µg ethinyl estradiol (estrogen component) and 1 mg norethindrone (progestin component), in obese otherwise healthy volunteers, decreased the exposure of ethinyl estradiol by 16% and increased the exposure of norethindrone by 22% [see Clinical Pharmacology (12.3)] . Although this interaction is not anticipated to increase the risk of pregnancy, irregular bleeding (spotting) may occur more frequently due to both the increased exposure to the progestin and lower exposure to the estrogen, which tends to stabilize the endometrium. |
| Intervention | Inform patients not to discontinue their combination oral contraceptive if spotting occurs, but to notify their healthcare provider if the spotting is troubling to them. |
| CNS Depressants Including Alcohol | |
| Clinical Impact | The concomitant use of alcohol or CNS depressant drugs (e.g., barbiturates, benzodiazepines, and sleep medications) with phentermine or topiramate may potentiate CNS depression such as dizziness or cognitive adverse reactions, or other centrally mediated effects of these agents. |
| Intervention | Advise patients not to drive or operate machinery until they have gained sufficient experience on QSYMIA to gauge whether it adversely affects their mental performance, motor performance, and/or vision. Caution patients against excessive alcohol intake when taking QSYMIA. Consider QSYMIA dosage reduction or discontinuation if cognitive dysfunction persists. [see Warnings and Precautions (5.6)] . |
| Non-Potassium Sparing Diuretics | |
| Clinical Impact | Concurrent use of QSYMIA with non-potassium sparing diuretics may potentiate the potassium-wasting action of these diuretics. Concomitant administration of hydrochlorothiazide alone with topiramate alone has been shown to increase the C maxand AUC of topiramate by 27% and 29%, respectively. |
| Intervention | When QSYMIA is used concomitantly with non-potassium-sparing diuretics, measure potassium before and during QSYMIA treatment [see Warnings and Precautions (5.15)and Clinical Pharmacology (12.3)] . |
| Antiepileptic Drugs | |
| Clinical Impact | Concomitant administration of phenytoin or carbamazepine with topiramate in patients with epilepsy, decreased plasma concentrations of topiramate by 48% and 40%, respectively, when compared to topiramate given alone [see Clinical Pharmacology (12.3)] . Concomitant administration of valproic acid and topiramate has been associated with hyperammonemia with and without encephalopathy. Concomitant administration of topiramate with valproic acid in patients has also been associated with hypothermia (with and without hyperammonemia). |
| Intervention | Consider measuring blood ammonia in patients in whom the onset of hypothermia or encephalopathy has been reported [see Clinical Pharmacology (12.3)] . |
| Carbonic Anhydrase Inhibitors | |
| Clinical Impact | Concomitant use of topiramate with any other carbonic anhydrase inhibitor may increase the severity of metabolic acidosis and may also increase the risk of kidney stone formation. |
| Intervention | Avoid the use of QSYMIA with other drugs that inhibit carbonic anhydrase. If concomitant use of QSYMIA with another carbonic anhydrase inhibitor is unavoidable, monitor patient for the appearance or worsening of metabolic acidosis [see Warnings and Precautions (5.8, 5.13)] . |
| Pioglitazone | |
| Clinical Impact | A decrease in the exposure of pioglitazone and its active metabolites were noted with the concurrent use of pioglitazone and topiramate in a clinical trial. The clinical relevance of these observations is unknown. |
| Intervention | Consider increased glycemic monitoring when using pioglitazone and QSYMIA concomitantly [see Clinical Pharmacology (12.3)]. |
| Amitriptyline | |
| Clinical Impact | Some patients may experience a large increase in amitriptyline concentration in the presence of topiramate. |
| Intervention | Any adjustments in amitriptyline dose when used with QSYMIA should be made according to the patient's clinical response and not on the basis of amitriptyline levels [see Clinical Pharmacology (12.3)] . |
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
QSYMIA is contraindicated in pregnant patients. The use of QSYMIA can cause fetal harm, and weight loss offers no clear clinical benefit to a pregnant patient (see Clinical Considerations) . Available data from pregnancy registries and epidemiologic studies indicate an increased risk of major congenital malformations, including but not limited to cleft lip and/or cleft palate (oral clefts), and of being SGA in infants exposed in uteroto topiramate (see Data) . When phentermine and topiramate were co-administered to rats at doses of 3.75 and 25 mg/kg, respectively [approximately 2 times the maximum recommended human dose (MRHD) based on area under the curve (AUC)], or at the same dose to rabbits (approximately 0.1 times and 1 time, respectively, the clinical exposures at the MRHD based on AUC), there were no drug-related malformations. However, structural malformations, including craniofacial defects and reduced fetal weights occurred in offspring of multiple species of pregnant animals administered topiramate at clinically relevant doses (see Data) . Advise pregnant women of the potential risk to a fetus.
Clinical Considerations
Disease Associated Maternal and/or Embryo/Fetal Risk
Weight loss during pregnancy may result in fetal harm. Appropriate weight gain based on pre-pregnancy weight is currently recommended for all pregnant patients, including those who are already overweight or obese, due to the obligatory weight gain that occurs in maternal tissues during pregnancy. Maternal obesity increases the risk for congenital malformations, including neural tube defects, cardiac malformations, oral clefts, and limb reduction defects.
Fetal/Neonatal Adverse Reactions
QSYMIA can cause metabolic acidosis [see Warnings and Precautions (5.8)] . The effect of topiramate-induced metabolic acidosis has not been studied in pregnancy; however, metabolic acidosis in pregnancy (due to other causes) can cause decreased fetal growth, decreased fetal oxygenation, and fetal death, and may affect the fetus' ability to tolerate labor.
Data
Human Data
Data evaluating the risk of major congenital malformations, oral clefts, and being SGA with topiramate exposure during pregnancy is available from the North American Antiepileptic Drug (NAAED) Pregnancy Registry and from several larger retrospective epidemiologic studies.
Major Congenital Malformations
The NAAED Pregnancy Registry indicates an increased risk of major congenital malformations, including but not limited to oral clefts in infants exposed to topiramate during the first trimester of pregnancy. Other than oral clefts, no specific pattern of major congenital malformations or grouping of major congenital malformation types were observed. In the NAAED pregnancy registry, when topiramate-exposed infants with only oral clefts were excluded, the prevalence of major congenital malformations (4.1%) was higher than that in infants exposed to a reference antiepileptic drug (AED) (1.8%) or in infants with mothers without epilepsy and without exposure to AEDs (1.1%).
Oral Clefts
In the NAAED Pregnancy Registry, the prevalence of oral clefts among topiramate-exposed infants (1.4%) was higher than the prevalence in infants exposed to a reference AED (0.3%) or the prevalence in infants with mothers without epilepsy and without exposure to AEDs (0.11%). It was also higher than the background prevalence in United States (0.17%) as estimated by the Centers for Disease Control and Prevention (CDC). The relative risk of oral clefts in topiramate-exposed pregnancies in the NAAED Pregnancy Registry was 12.5 (95% Confidence Interval [CI] 5.9-26.37) as compared to the risk in a background population of untreated women. The UK Epilepsy and Pregnancy Register reported a prevalence of oral clefts among infants exposed to topiramate monotherapy (3.2%) that was 16 times higher than the background rate in the UK (0.2%).
Larger retrospective epidemiology studies showed that topiramate monotherapy exposure in pregnancy is associated with an approximately two to five-fold increased risk of oral clefts. The FORTRESS study found an excess risk of 1.5 (95% CI = -1.1 to 4.1) oral cleft cases per 1,000 infants exposed to topiramate during the first trimester.
Small for Gestational Age
Data from the NAAED Pregnancy Registry and population-based birth registry cohort indicate that exposure to topiramate in utero is associated with an increased risk of SGA newborns (birth weight <10 thpercentile). In the NAAED Pregnancy Registry, 19.7% of topiramate-exposed newborns were SGA compared to 7.9% of newborns exposed to a reference AED and 5.4% of newborns of mothers without epilepsy and without AED exposure. In the medical Birth Registry of Norway, a population-based pregnancy registry, 25% of newborns in the topiramate monotherapy exposure group were SGA compared to 9% in the comparison group unexposed to AEDs. The long-term consequences of the SGA findings are not known.
Animal Data
Phentermine/Topiramate
Embryo-fetal development studies have been conducted in rats and rabbits with combination phentermine and topiramate treatment. Phentermine and topiramate co-administered to rats during the period of organogenesis (gestation day (GD) 6 through 17) caused reduced fetal body weights but did not cause fetal malformations at the maximum dose of 3.75 mg/kg phentermine and 25 mg/kg topiramate [approximately 2 times the maximum recommended human dose (MRHD) based on area under the curve (AUC) estimates for each active ingredient]. In a similar study in rabbits in which the same doses were administered from GD 6 through 18, no effects on embryo-fetal development were observed at approximately 0.1 times (phentermine) and 1 time (topiramate) clinical exposures at the MRHD based on AUC. Significantly lower maternal body weight gain was recorded at these doses in rats and rabbits.
A pre- and post-natal development study was conducted in rats with combination phentermine and topiramate treatment. There were no adverse maternal or offspring effects in rats treated throughout organogenesis and lactation with 1.5 mg/kg/day phentermine and 10 mg/kg/day topiramate (approximately 2- and 3-times clinical exposures at the MRHD, respectively, based on AUC). Treatment with higher doses of 11.25 mg/kg/day phentermine and 75 mg/kg/day topiramate (approximately 5 and 6 times maximum clinical doses based on AUC, respectively) caused reduced maternal body weight gain and offspring toxicity. Offspring effects included lower pup survival after birth, increased limb and tail malformations, reduced pup body weight and delayed growth, development, and sexual maturation without affecting learning, memory, or fertility and reproduction. The limb and tail malformations were consistent with results of animal studies conducted with topiramate alone.
Phentermine
Animal reproduction studies have not been conducted with phentermine. Limited data from studies conducted with the phentermine/topiramate combination indicate that phentermine alone was not teratogenic but resulted in lower body weight and reduced survival of offspring in rats at 5-fold the MRHD of QSYMIA, based on AUC.
Topiramate
Topiramate causes developmental toxicity, including teratogenicity, at clinically relevant doses in multiple animal species.
Developmental toxicity, including teratogenicity, occurred at clinically relevant doses in multiple animal species in which topiramate was administered during the period of organogenesis (GD 6 – 15 in rodents, GD 6 – 18 in rabbits. In these studies, fetal malformations (primarily craniofacial defects such as cleft palate), limb malformations (ectrodactyly, micromelia, and amelia), rib/vertebral column anomalies, and/or reduced fetal weights were observed at dosages ≥ 20 mg/kg in mice (approximately 2 times the MRHD of topiramate in QSYMIA 15 mg/92 mg on a mg/m 2basis), 20 mg/kg in rats (2 times the MRHD of QSYMIA based on estimated AUC), and 35 mg/kg in rabbits (2 times the MRHD based on estimated AUC). When rats were administered topiramate from GD 15 through lactation day 20, reductions in pre- and/or post-weaning weights occurred at dosages ≥ 2 mg/kg (2 times the MRHD of QSYMIA based on estimated AUC).
8.2 Lactation
Risk Summary
Topiramate and phentermine are present in human milk. There are no data on the effects of topiramate and phentermine on milk production. Diarrhea and somnolence have been reported in breastfed infants with maternal use of topiramate. There are no data on the effects of phentermine in breastfed infants. Because of the potential for serious adverse reactions, including changes in sleep, irritability, hypertension, vomiting, tremor, and weight loss in breastfed infants with maternal use of phentermine, advise patients that breastfeeding is not recommended during QSYMIA therapy.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy testing is recommended in patients who can become pregnant before initiating QSYMIA and monthly during QSYMIA therapy [see Warnings and Precautions (5.1), Use in Specific Populations (8.1)] .
Contraception
Females
QSYMIA can cause fetal harm when administered to a pregnant patient [see Use in Specific Populations (8.1)]. Advise patients who can become pregnant to use effective contraception during therapy with QSYMIA.
For patients taking combined oral contraceptives (COCs), use of QSYMIA may cause irregular bleeding [see Drug Interactions (7)] . Advise patients not to discontinue taking their COC and to contact their healthcare provider.
8.4 Pediatric Use
The safety and effectiveness of QSYMIA as an adjunct to a reduced-calorie diet and increased physical activity for chronic weight management in pediatric patients aged 12 years and older with a BMI in the 95th percentile or greater standardized for age and sex have been established. Use of QSYMIA for this indication is supported by a 56-week, double-blind, placebo-controlled study in 223 pediatric patients aged 12 years and above, a pharmacokinetic study in pediatric patients, and studies in adults with obesity [see Clinical Pharmacology (12.3)and Clinical Studies (14)] .
In a pediatric clinical trial, there was one episode of serious suicidal ideation in a QSYMIA-treated patient requiring hospitalization and pharmacologic treatment [see Warnings and Precautions (5.3)] ; more patients treated with QSYMIA versus placebo reported adverse reactions related to mood (e.g., depression, anxiety) and sleep disorders (e.g., insomnia) [see Warnings and Precautions (5.5)] . Increases in bone mineral density and linear growth were attenuated in QSYMIA- versus placebo-treated patients [see Warnings and Precautions (5.7)] . Serious adverse reactions seen in pediatric patients using topiramate include acute angle glaucoma, oligohidrosis and hyperthermia, metabolic acidosis, cognitive and neuropsychiatric reactions, hyperammonemia and encephalopathy, and kidney stones.
The safety and effectiveness of QSYMIA in pediatric patients below the age of 12 years have not been established.
8.5 Geriatric Use
In the QSYMIA clinical trials, a total of 254 (7%) of the patients were 65 to 69 years of age; no patients 70 years of age or older were enrolled.
Clinical studies of QSYMIA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Compared to healthy volunteers with normal renal function, patients with moderate and severe renal impairment as estimated by the Cockcroft-Gault equation had higher exposures to phentermine and topiramate.
The recommended dosage of QSYMIA in patients with mild renal impairment (CrCl greater or equal to 50 and less than 80 mL/min) is the same as the recommended dosage for patients with normal renal function.
In patients with moderate (CrCl greater than or equal to 30 to less than 50 mL/min) and severe (CrCl less than 30 mL/min) renal impairment, the maximum recommended dosage is QSYMIA 7.5 mg/46 mg once daily.
QSYMIA has not been studied in patients with end-stage renal disease on dialysis. Avoid QSYMIA in this patient population [see Dosage and Administration (2.5)and Clinical Pharmacology (12.3)] .
8.7 Hepatic Impairment
In patients with mild (Child-Pugh 5 - 6) and moderate (Child-Pugh 7 - 9) hepatic impairment, exposure to phentermine was higher compared to healthy volunteers with normal hepatic function. Exposure to topiramate was similar among patients with mild and moderate hepatic impairment and healthy volunteers.
The recommended dosage of QSYMIA in patients with mild hepatic impairment (Child-Pugh 5 - 6) is the same as the recommended dosage in patients with normal hepatic function.
In patients with moderate hepatic impairment, the maximum recommended dosage is QSYMIA 7.5 mg/46 mg once daily.
QSYMIA has not been studied in patients with severe hepatic impairment (Child-Pugh score 10 - 15). Avoid QSYMIA in this patient population [see Dosage and Administration (2.6)and Clinical Pharmacology (12.3)] .
9. Drug Abuse and Dependence
9.1 Controlled Substance
QSYMIA contains phentermine, a Schedule IV controlled substance, and topiramate, which is not a controlled substance.
9.2 Abuse
Phentermine has a known potential for abuse. Abuse is the intentional, non-therapeutic use of a drug, even once, for its desirable psychological or physiological effects.
Phentermine is related chemically and pharmacologically to amphetamines. Amphetamines and other stimulant drugs have been extensively abused. Abuse of amphetamines and related drugs (e.g., phentermine) may be associated with impaired control over drug use and severe social dysfunction. There are reports of patients who have increased the dosage of these drugs to many times higher than recommended. Assess the risk of abuse prior to prescribing QSYMIA as part of a chronic weight management program.
9.3 Dependence
Physical dependence may occur in patients treated with QSYMIA. Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms after abrupt discontinuation or a significant dose reduction of a drug.
The following adverse reactions have been associated with the abrupt discontinuation of the individual components of QSYMIA:
- For topiramate, abrupt discontinuation has been associated with seizures in patients without a history of seizures or epilepsy [see Warnings and Precautions (5.12)] .
- For phentermine, abrupt discontinuation following prolonged high dosage administration results in extreme fatigue and mental depression; changes are also noted on a sleep electroencephalogram.
Thus, in situations where rapid withdrawal of QSYMIA is required, appropriate medical monitoring is recommended. Patients discontinuing QSYMIA 15 mg/92 mg should be gradually tapered to reduce the possibility of precipitating a seizure [see Dosage and Administration (2.4)] .
10. Overdosage
In the event of a significant overdose with QSYMIA, if the ingestion is recent, the stomach should be emptied immediately by gastric lavage or by induction of emesis. Appropriate supportive treatment should be provided according to the patient's clinical signs and symptoms.
Acute overdose of phentermine may be associated with restlessness, tremor, hyperreflexia, rapid respiration, confusion, aggressiveness, hallucinations, and panic states. Fatigue and depression usually follow the central stimulation. Cardiovascular effects include arrhythmia, hypertension or hypotension, and circulatory collapse. Gastrointestinal symptoms include nausea, vomiting, diarrhea, and abdominal cramps. Fatal poisoning usually terminates in convulsions and coma. Manifestations of chronic intoxication with anorectic drugs include severe dermatoses, marked insomnia, irritability, hyperactivity, and personality changes. A severe manifestation of chronic intoxication is psychosis, often clinically indistinguishable from schizophrenia.
Management of acute phentermine intoxication is largely symptomatic and includes lavage and sedation with a barbiturate. Acidification of the urine increases phentermine excretion. Intravenous phentolamine has been suggested for possible acute, severe hypertension, if this complicates phentermine overdosage.
Topiramate overdose has resulted in severe metabolic acidosis. Other signs and symptoms include convulsions, drowsiness, speech disturbance, blurred vision, diplopia, impaired mentation, lethargy, abnormal coordination, stupor, hypotension, abdominal pain, agitation, dizziness, and depression. The clinical consequences were not severe in most cases, but deaths have been reported after overdoses involving topiramate. A patient who ingested a dose between 96 and 110 gm topiramate was admitted to hospital with coma lasting 20 to 24 hours followed by full recovery after 3 to 4 days.
Hemodialysis is an effective means of removing topiramate from the body.
11. Qsymia Description
QSYMIA extended-release capsules are comprised of immediate-release phentermine hydrochloride (expressed as the weight of the free base) and extended-release topiramate. QSYMIA contains phentermine hydrochloride, a sympathomimetic amine anorectic, and topiramate, a sulfamate-substituted monosaccharide.
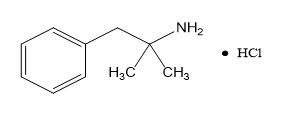
Phentermine Hydrochloride
The chemical name of phentermine hydrochloride is α,α-dimethylphenethylamine hydrochloride. The molecular formula is C 10H 15N ∙ HCl and its molecular weight is 185.7 (hydrochloride salt) or 149.2 (free base). Phentermine hydrochloride is a white, odorless, hygroscopic, crystalline powder that is soluble in water, methanol, and ethanol. Its structural formula is:
Topiramate
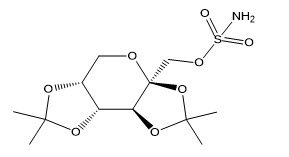
Topiramate is 2,3:4,5-di-O-isopropylidene-β-D-fructopyranose sulfamate. The molecular formula is C 12H 21NO 8S and its molecular weight is 339.4. Topiramate is a white to off-white crystalline powder with a bitter taste. It is freely soluble in methanol and acetone, sparingly soluble in pH 9 to pH 12 aqueous solutions and slightly soluble in pH 1 to pH 8 aqueous solutions. Its structural formula is:
QSYMIA
QSYMIA (phentermine and topiramate extended-release capsules) is for oral administration and available in four dosage strengths:
- 3.75 mg/23 mg (phentermine 3.75 mg and topiramate 23 mg) (equivalent to 4.67 mg of Phentermine Hydrochloride USP).
- 7.5 mg/46 mg (phentermine 7.5 mg and topiramate 46 mg) (equivalent to 9.33 mg of Phentermine Hydrochloride USP).
- 11.25 mg/69 mg (phentermine 11.25 mg and topiramate 69 mg) (equivalent to 14.0 mg of Phentermine Hydrochloride USP).
- 15 mg/92 mg (phentermine 15 mg and topiramate 92 mg) (equivalent to 18.66 mg of Phentermine Hydrochloride USP).
Each capsule contains the following inactive ingredients: FD&C Blue #1, FD&C Red #3, FD&C Yellow #5 and #6, ethylcellulose, gelatin, methylcellulose, microcrystalline cellulose, povidone, starch, sucrose, talc, titanium dioxide, and pharmaceutical black and white inks.
12. Qsymia - Clinical Pharmacology
12.1 Mechanism of Action
Phentermine is a sympathomimetic amine with pharmacologic activity similar to the prototype drugs of this class used in obesity, amphetamine (d- and d/l-amphetamine). Drugs of this class used in obesity are commonly known as "anorectics" or "anorexigenics." The effect of phentermine on chronic weight management is likely mediated by release of catecholamines in the hypothalamus, resulting in reduced appetite and decreased food consumption, but other metabolic effects may also be involved. The exact mechanism of action is not known.
The precise mechanism of action of topiramate on chronic weight management is not known. Topiramate's effect on chronic weight management may be due to its effects on both appetite suppression and satiety enhancement, induced by a combination of pharmacologic effects including augmenting the activity of the neurotransmitter gamma-aminobutyrate, modulation of voltage-gated ion channels, inhibition of AMPA/kainite excitatory glutamate receptors, or inhibition of carbonic anhydrase.
12.2 Pharmacodynamics
Typical actions of amphetamines include central nervous system stimulation and elevation of blood pressure. Tachyphylaxis and tolerance have been demonstrated with drugs in this class.
Cardiac Electrophysiology
The effect of QSYMIA on the QTc interval was evaluated in a randomized, double-blind, placebo- and active-controlled (400 mg moxifloxacin), and parallel group/crossover thorough QT/QTc study. A total of 54 healthy subjects were administered QSYMIA 7.5 mg/46 mg at steady state and then titrated to QSYMIA 22.5 mg/138 mg at steady state. QSYMIA 22.5 mg/138 mg [a supra-therapeutic dose resulting in a phentermine and topiramate maximum concentration (C max) of 4- and 3- times higher than those at QSYMIA 7.5 mg/46 mg, respectively] did not affect cardiac repolarization as measured by the change from baseline in QTc.
Glomerular Filtration Rate (GFR)
Healthy obese men and women received QSYMIA daily for 4 weeks (3.75 mg/23 mg on Days 1 to 3, 7.5 mg/46 mg on Days 4 to 6, 11.25 mg/69 mg on Days 7 to 9, and 15 mg/92 mg on Days 10 to 28). The glomerular filtration rate (GFR) of these participants was assessed via iohexol clearance. On average, GFR decreased during QSYMIA treatment and returned to baseline within 4 weeks after discontinuing QSYMIA [see Warnings and Precautions (5.9)]
12.3 Pharmacokinetics
Absorption
Phentermine
Upon oral administration of a single QSYMIA 15 mg/92 mg, the resulting mean plasma phentermine maximum concentration (C max), time to C max(T max), area under the concentration curve from time zero to the last time with measurable concentration (AUC 0-t), and area under the concentration curve from time zero to infinity (AUC 0-∞) are 49.1 ng/mL, 6 hr, 1990 ng∙hr/mL, and 2000 ng∙hr/mL, respectively. A high fat meal does not affect phentermine pharmacokinetics for QSYMIA 15 mg/92 mg. Phentermine pharmacokinetics are approximately dose-proportional from QSYMIA 3.75 mg/23 mg to phentermine 15 mg/topiramate 100 mg. Upon dosing phentermine 15 mg/topiramate 100 mg fixed dose combination capsule to steady state, the mean phentermine accumulation ratios for AUC and C maxare both approximately 2.5.
Topiramate
Upon oral administration of a single QSYMIA 15 mg/92 mg, the resulting mean plasma topiramate C max, T max, AUC 0-t, and AUC 0-∞, are 1020 ng/mL, 9 hr, 61600 ng∙hr/mL, and 68000 ng∙hr/mL, respectively. A high fat meal does not affect topiramate pharmacokinetics for QSYMIA 15 mg/92 mg. Topiramate pharmacokinetics are approximately dose-proportional from QSYMIA 3.75 mg/23 mg to phentermine 15 mg/topiramate 100 mg. Upon dosing phentermine 15 mg/topiramate 100 mg fixed dose combination capsule to steady state, the mean topiramate accumulation ratios for AUC and C maxare both approximately 4.0.
Distribution
Phentermine
Phentermine is 17.5% plasma protein bound. The estimated phentermine apparent volume of distribution (Vd/F) is 348 L via population pharmacokinetic analysis.
Topiramate
Topiramate is 15 - 41% plasma protein bound over the blood concentration range of 0.5 to 250 µg/mL. The fraction bound decreased as blood topiramate increased. The estimated topiramate Vc/F (volume of the central compartment), and Vp/F (volume of the peripheral compartment) are 50.8 L, and 13.1 L, respectively, via population pharmacokinetic analysis.
Elimination
Metabolism and Excretion
Phentermine
Phentermine has two metabolic pathways, namely p-hydroxylation on the aromatic ring and N-oxidation on the aliphatic side chain. Cytochrome P450 (CYP) 3A4 primarily metabolizes phentermine but does not show extensive metabolism. Monoamine oxidase (MAO)-A and MAO-B do not metabolize phentermine. Seventy to 80% of a dose exists as unchanged phentermine in urine when administered alone. The mean phentermine terminal half-life is about 20 hours. The estimated phentermine oral clearance (CL/F) is 8.79 L/h via population pharmacokinetic analysis.
Topiramate
Topiramate does not show extensive metabolism. Six topiramate metabolites (via hydroxylation, hydrolysis, and glucuronidation) exist, none of which constitutes more than 5% of an administered dose. About 70% of a dose exists as unchanged topiramate in urine when administered alone. The mean topiramate terminal half-life is about 65 hours. The estimated topiramate CL/F is 1.17 L/h via population pharmacokinetic analysis.
Specific Populations
Patients with Renal Impairment
A single-dose, open-label study was conducted to evaluate the pharmacokinetics of QSYMIA 15 mg/92 mg in adult patients with varying degrees of chronic renal impairment compared to healthy volunteers with normal renal function. The study included patients with renal impairment classified on the basis of creatinine clearance as mild (greater or equal to 50 and less than 80 mL/min), moderate (greater than or equal to 30 and less than 50 mL/min), and severe (less than 30 mL/min). Creatinine clearance was estimated from serum creatinine based on the Cockcroft-Gault equation.
Compared to healthy volunteers, phentermine AUC 0-infwas 91%, 45%, and 22% higher in patients with severe, moderate, and mild renal impairment, respectively; phentermine C maxwas 2% to 15% higher. Compared to healthy volunteers, topiramate AUC 0-infwas 126%, 85%, and 25% higher for patients with severe, moderate, and mild renal impairment, respectively; topiramate C maxwas 6% to 17% higher. An inverse relationship between phentermine or topiramate C maxor AUC and creatinine clearance was observed.
QSYMIA has not been studied in patients with end-stage renal disease on dialysis [see Dosage and Administration (2.5), and Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
A single-dose, open-label study was conducted to evaluate the pharmacokinetics of QSYMIA 15 mg/92 mg in healthy volunteers with normal hepatic function compared with patients with mild (Child-Pugh score 5 - 6) and moderate (Child-Pugh score 7 - 9) hepatic impairment. In patients with mild and moderate hepatic impairment, phentermine AUC was 37% and 60% higher compared to healthy volunteers. Pharmacokinetics of topiramate was not affected in patients with mild and moderate hepatic impairment when compared with healthy volunteers. QSYMIA has not been studied in patients with severe hepatic impairment (Child-Pugh score 10 - 15) [see Dosage and Administration (2.6), and Use in Specific Populations (8.7)] .
Pediatric Patients 12 to 17 years old
A randomized, double-blind, placebo-controlled study was conducted to evaluate the population pharmacokinetics of QSYMIA using data from 37 pediatric patients (12 to 17 years of age) with obesity. QSYMIA dosages of 3.75 mg/23 mg, 7.5 mg/46 mg, and 15 mg/92 mg were studied. QSYMIA exposure in the pediatric patients appeared comparable to that in adults.
Drug Interaction Studies
In Vitro Assessment of Drug Interactions
Effects of Phentermine/Topiramate on Other Drugs
Table 8 describes the effect of phentermine/topiramate on the pharmacokinetics of co-administered drugs.
| Phentermine/Topiramate | Co-administered Drug and Dosing Regimen | ||
|---|---|---|---|
| Drug and Dose (mg) | Change in AUC | Change in C max | |
|
|||
| *15 mg/92 mg dose QD for 16 days | Metformin 500 mg BID for 5 days | ↑ 23% | ↑ 16% |
| *15 mg/92 mg dose QD for 21 days | Sitagliptin 100 mg QD for 5 days | ↓ 3% | ↓ 9% |
| †15 mg/92 mg dose QD for 15 days | Oral contraceptive single dose
norethindrone 1 mg ethinyl estradiol 35 mcg | ↑ 16%
↓ 16% | ↑ 22%
↓ 8% |
Effect of Other Drugs on Phentermine/Topiramate
Table 9 describes the effect of other drugs on the pharmacokinetics of phentermine/topiramate.
| Co-administered Drug and Dosing Regimen | Phentermine/Topiramate | ||
|---|---|---|---|
| Dose (mg) | Change in AUC | Change in C max | |
|
|||
| Topiramate 92 mg single dose | 15 mg phentermine single dose | ↑ 42% | ↑ 13% |
| Phentermine 15 mg single dose | 92 mg topiramate single dose | ↑ 6% | ↑ 2% |
| *Metformin 500 mg BID for 5 days | 15 mg/92 mg dose QD for 16 days
phentermine topiramate | ↑ 5%
↓ 5% | ↑ 7%
↓ 4% |
| *Sitagliptin 100 mg QD for 5 days | 15 mg/92 mg dose QD for 21 days
phentermine topiramate | ↑ 9%
↓ 2% | ↑ 10%
↓ 2% |
| *Probenecid 2 g QD | 15 mg/92 mg dose QD for 11 days
phentermine topiramate | ↓ 0.3%
↑ 0.7% | ↑ 4%
↑ 3% |
Effects of Topiramate Alone on Other Drugs and Effects of Other Drugs on Topiramate
Antiepileptic Drugs
Potential interactions between topiramate and standard antiepileptic (AED) drugs were assessed in controlled clinical pharmacokinetic studies in patients with epilepsy. The effects of these interactions on mean plasma AUCs are summarized in Table 10.
In Table 10, the second column (AED concentration) describes what happens to the concentration of the AED listed in the first column when topiramate is added. The third column (topiramate concentration) describes how the co-administration of a drug listed in the first column modifies the concentration of topiramate in experimental settings when topiramate was given alone [see Drug Interactions (7)] .
| AED
Co-administered | AED
Concentration | Topiramate
Concentration |
|---|---|---|
| NC = Less than 10% change in plasma concentration; NE = Not Evaluated; TPM = topiramate | ||
| Phenytoin | NC or 25% increase * | 48% decrease |
| Carbamazepine (CBZ) | NC | 40% decrease |
| CBZ epoxide † | NC | NE |
| Valproic acid | 11% decrease | 14% decrease |
| Phenobarbital | NC | NE |
| Primidone | NC | NE |
| Lamotrigine | NC at TPM doses up to 400 mg/day | 13% decrease |
Digoxin
In a single-dose study, serum digoxin AUC was decreased by 12% with concomitant topiramate administration. The clinical relevance of this observation has not been established.
Hydrochlorothiazide
A drug-drug interaction study conducted in healthy volunteers evaluated the steady-state pharmacokinetics of hydrochlorothiazide (HCTZ) (25 mg q24h) and topiramate (96 mg q12h) when administered alone and concomitantly. The results of this study indicate that topiramate C maxincreased by 27% and AUC increased by 29% when HCTZ was added to topiramate. The clinical significance of this change is unknown. The steady-state pharmacokinetics of HCTZ were not significantly influenced by the concomitant administration of topiramate. Clinical laboratory results indicated decreases in serum potassium after topiramate or HCTZ administration, which were greater when HCTZ and topiramate were administered in combination [see Drug Interactions (7)and Warnings and Precautions (5.15)] .
Pioglitazone
A drug-drug interaction study conducted in healthy volunteers evaluated the steady-state pharmacokinetics of topiramate (96 mg twice daily) and pioglitazone (30 mg daily) when administered alone and concomitantly for 7 days. A 15% decrease in the area under the concentration-time curve during a dosage interval at steady state (AUC τ,ss) of pioglitazone with no alteration in maximum steady-state plasma drug concentration during a dosage interval (C max,ss) was observed. This finding was not statistically significant. In addition, a 13% and 16% decrease in C max,ssand AUC τ,ssrespectively, of the active hydroxy-metabolite was noted as well as a 60% decrease in C max,ssand AUC τ,ssof the active keto-metabolite [see Drug Interactions (7)] .
Glyburide
A drug-drug interaction study conducted in patients with type 2 diabetes evaluated the steady-state pharmacokinetics of glyburide (5 mg/day) alone and concomitantly with topiramate (150 mg/day). There was a 22% decrease in C maxand a 25% reduction in AUC 24for glyburide during topiramate administration. Systemic exposure (AUC) of the active metabolites, 4- trans-hydroxyglyburide (M1), and 3- cis-hydroxyglyburide (M2), was reduced by 13% and 15%, and C maxwas reduced by 18% and 25%, respectively. The steady-state pharmacokinetics of topiramate were unaffected by concomitant administration of glyburide.
Lithium
In patients, the pharmacokinetics of lithium were unaffected during treatment with topiramate at doses of 200 mg/day; however, there was an observed increase in systemic exposure of lithium (27% for C maxand 26% for AUC) following topiramate doses up to 600 mg/day.
Haloperidol
The pharmacokinetics of a single dose of haloperidol (5 mg) were not affected following multiple dosing of topiramate (100 mg every 12 hours) in 13 healthy adults (6 males, 7 females).
Amitriptyline
There was a 12% increase in AUC and C maxfor amitriptyline (25 mg per day) in 18 normal subjects (9 males, 9 females) receiving 200 mg/day of topiramate [see Drug Interactions (7)] .
Sumatriptan
Multiple dosing of topiramate (100 mg every 12 hrs) in 24 healthy volunteers (14 males, 10 females) did not affect the pharmacokinetics of single-dose sumatriptan either orally (100 mg) or subcutaneously (6 mg).
Risperidone
When administered concomitantly with topiramate at escalating doses of 100, 250, and 400 mg/day, there was a reduction in risperidone systemic exposure (16% and 33% for steady-state AUC at the 250 and 400 mg/day doses of topiramate). No alterations of 9-hydroxyrisperidone levels were observed. Co-administration of topiramate 400 mg/day with risperidone resulted in a 14% increase in C maxand a 12% increase in AUC 12of topiramate. There were no clinically significant changes in the systemic exposure of risperidone plus 9-hydroxyrisperidone or of topiramate; therefore, this interaction is not likely to be of clinical significance.
Propranolol
Multiple dosing of topiramate (200 mg/day) in 34 healthy volunteers (17 males, 17 females) did not affect the pharmacokinetics of propranolol following daily 160 mg doses. Propranolol doses of 160 mg/day in 39 volunteers (27 males, 12 females) had no effect on the exposure to topiramate, at a dose of 200 mg/day of topiramate.
Dihydroergotamine
Multiple dosing of topiramate (200 mg/day) in 24 healthy volunteers (12 males, 12 females) did not affect the pharmacokinetics of a 1 mg subcutaneous dose of dihydroergotamine. Similarly, a 1 mg subcutaneous dose of dihydroergotamine did not affect the pharmacokinetics of a 200 mg/day dose of topiramate in the same study.
Diltiazem
Co-administration of diltiazem (240 mg Cardizem CD ®) with topiramate (150 mg/day) resulted in a 10% decrease in C maxand a 25% decrease in diltiazem AUC, a 27% decrease in C maxand an 18% decrease in des-acetyl diltiazem AUC, and no effect on N-desmethyl diltiazem. Co-administration of topiramate with diltiazem resulted in a 16% increase in C maxand a 19% increase in AUC 12of topiramate.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Phentermine/Topiramate
No animal studies have been conducted with the combination of phentermine/topiramate to evaluate carcinogenesis, mutagenesis, or impairment of fertility. The following data are based on findings in studies performed individually with phentermine or topiramate, QSYMIA's two active ingredients.
Phentermine
Phentermine was not mutagenic or clastogenic with or without metabolic activation in the Ames bacterial mutagenicity assay, a chromosomal aberration test in Chinese hamster lung (CHL-K1) cells, or an in vivomicronucleus assay.
Rats were administered oral doses of 3, 10, and 30 mg/kg/day phentermine for 2 years. There was no evidence of carcinogenicity at the highest dose of phentermine (30 mg/kg) which is approximately 11 to 15 times the maximum recommended clinical dose of QSYMIA 15 mg/92 mg based on AUC exposure.
No animal studies have been conducted with phentermine to determine the potential for impairment of fertility.
Topiramate
Topiramate did not demonstrate genotoxic potential when tested in a battery of in vitroand in vivoassays. Topiramate was not mutagenic in the Ames test or the in vitromouse lymphoma assay; it did not increase unscheduled DNA synthesis in rat hepatocytes in vitro; and it did not increase chromosomal aberrations in human lymphocytes in vitroor in rat bone marrow in vivo.
An increase in urinary bladder tumors was observed in mice given topiramate (20, 75, and 300 mg/kg) in the diet for 21 months. The elevated bladder tumor incidence, which was statistically significant in males and females receiving 300 mg/kg, was primarily due to the increased occurrence of a smooth muscle tumor considered histomorphologically unique to mice. Plasma exposures in mice receiving 300 mg/kg were approximately 2 to 4 times steady-state exposures measured in patients receiving topiramate monotherapy at the MRHD of QSYMIA 15 mg/92 mg. The relevance of this finding to human carcinogenic risk is uncertain. No evidence of carcinogenicity was seen in rats following oral administration of topiramate for 2 years at doses up to 120 mg/kg (approximately 4 to 10 times the MRHD of QSYMIA based on AUC estimates).
No adverse effects on male or female fertility were observed in rats at doses up to 100 mg/kg (approximately 4 to 8 times male and female MRHD exposures of QSYMIA based on AUC).
14. Clinical Studies
Clinical Studies in Adults
The effect of QSYMIA on weight loss in conjunction with reduced caloric intake and increased physical activity was studied in two randomized, double-blind, placebo-controlled studies in patients with obesity (Study 1) and patients with obesity or overweight with two or more significant co-morbidities (Study 2). Both studies had a 4-week titration period, followed by 52 weeks of treatment. There were two co-primary efficacy outcomes measured after 1 year of treatment (Week 56): 1) the percent weight loss from baseline; and 2) treatment response defined as achieving at least 5% weight loss from baseline.
In Study 1, patients with obesity (BMI greater than or equal to 35 kg/m 2) were randomized to receive 1 year of treatment with placebo (N=514), QSYMIA 3.75 mg/23 mg (N=241), or QSYMIA 15 mg/92 mg (N=512) in a 2:1:2 ratio. Patients ranged in age from 18-71 years old (mean age 43) and 83% were female. Approximately 80% were Caucasian, 18% were African American, and 15% were Hispanic/Latino. At the beginning of the study the average weight and BMI of patients was 116 kg and 42 kg/m 2, respectively. Patients with type 2 diabetes were excluded from participating in Study 1. During the study, a well-balanced, reduced-calorie diet to result in an approximate 500 kcal/day decrease in caloric intake was recommended to all patients and patients were offered nutritional and lifestyle modification counseling.
In Study 2, patients with overweight or obesity were randomized to receive 1 year of treatment with placebo (N=994), QSYMIA 7.5 mg/46 mg (N=498), or QSYMIA 15 mg/92 mg (N=995) in a 2:1:2 ratio. Eligible patients had to have a BMI greater than or equal to 27 kg/m 2and less than or equal to 45 kg/m 2(there was no lower limit on BMI for patients with type 2 diabetes) and two or more of the following obesity-related co-morbid conditions:
- Elevated blood pressure (greater than or equal to 140/90 mmHg, or greater than or equal to 130/85 mmHg for diabetics) or requirement for greater than or equal to 2 antihypertensive medications;
- Triglycerides greater than 200-400 mg/dL or were receiving treatment with 2 or more lipid-lowering agents;
- Elevated fasting blood glucose (greater than 100 mg/dL) or diabetes; and/or
- Waist circumference greater than or equal to 102 cm for men or greater than or equal to 88 cm for women.
Patients ranged in age from 19-71 years old (mean age 51) and 70% were female. Approximately 86% were Caucasian, 12% were African American, and 13% were Hispanic/Latino. The average weight and BMI of patients at the start of the study was 103 kg and 36.6 kg/m 2, respectively. Approximately half (53%) of patients had hypertension at the start of the study. There were 388 (16%) patients with type 2 diabetes at the start of the study. During the study, a well-balanced, reduced-calorie diet to result in an approximate 500 kcal/day decrease in caloric intake was recommended to all patients and patients were offered nutritional and lifestyle modification counseling.
The percentage of randomized patients who withdrew from each study prior to week 56 was 40% in Study 1, and 31% in Study 2.
Table 11 provides the results for weight loss at 1 year in Studies 1 and 2. After 1 year of treatment with QSYMIA, all dose levels resulted in statistically significant weight loss compared to placebo (see Table 11, Figure 1and Figure 2). A statistically significant greater proportion of the patients randomized to QSYMIA than placebo achieved 5% and 10% weight loss.
| Study 1 (Obesity) | Study 2 (Obesity or Overweight with Co-morbidities) | |||||
|---|---|---|---|---|---|---|
| Analysis Method | Placebo | QSYMIA
3.75 mg/23 mg | QSYMIA
15 mg/92 mg | Placebo | QSYMIA
7.5 mg/46 mg | QSYMIA
15 mg/92 mg |
| SD=standard deviation; LS=least-squares; SE=standard error; CI=confidence interval | ||||||
| Type 1 error was controlled across all pairwise treatment comparisons. | ||||||
|
||||||
| ITT-LOCF (Primary) * | n = 498 | n = 234 | n = 498 | n = 979 | n = 488 | n = 981 |
| Weight (kg) | ||||||
| Baseline mean (SD) | 115.7 (21.4) | 118.6 (21.9) | 115.2 (20.8) | 103.3 (18.1) | 102.8 (18.2) | 103.1 (17.6) |
| % LS Mean Change from baseline (SE) † | -1.6 (0.4) | -5.1 (0.5) ‡ | -10.9 (0.4) ‡§ | -1.2 (0.3) | -7.8 (0.4) ‡ | -9.8 (0.3) ‡§ |
| Difference from placebo
(95% CI) | 3.5 (2.4-4.7) | 9.4 (8.4-10.3) | 6.6 (5.8-7.4) | 8.6 (8.0-9.3) | ||
| Percentage of patients losing greater than or equal to 5% body weight | 17% | 45% ‡ | 67% ‡§ | 21% | 62% ‡ | 70% ‡§ |
| Risk Difference vs. placebo
(95% CI) | 27.6 (20.4-34.8) | 49.4 (44.1-54.7) | 41.3 (36.3-46.3) | 49.2 (45.4-53.0) | ||
| Percentage of patients losing greater than or equal to 10% body weight | 7% | 19% ‡ | 47% ‡§ | 7% | 37% ‡ | 48% ‡§ |
| Risk Difference vs. placebo
(95% CI) | 11.4 (5.9-16.9) | 39.8 (34.8-44.7) | 29.9 (25.3-34.5) | 40.3 (36.7-43.8) | ||
| Figure 1. Study 1 Percent Weight Change from Baseline to Week 56 in Adults with Obesity | Figure 2. Study 2 Percent Weight Change from Baseline to Week 56 in Adults with Obesity or Overweight with Co-morbidities | ||
|---|---|---|---|
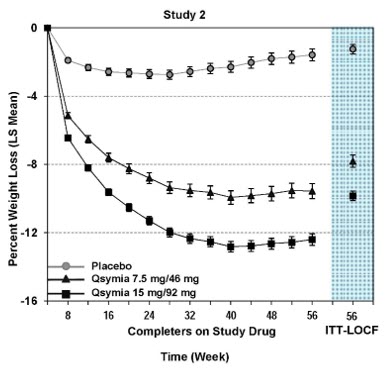 |
|||
The changes in cardiovascular, metabolic, and anthropometric risk factors associated with obesity from Study 1 and 2 are presented in Table 12 and Table 13. One year of therapy with QSYMIA resulted in relative improvement over placebo in several risk factors associated with obesity with the exception of heart rate [see Warnings and Precautions (5.2)] .
| Study 1 (Obesity) | Placebo
(N=498) | QSYMIA
3.75 mg/23 mg (N=234) | QSYMIA
15 mg/92 mg (N=498) | QSYMIA – Placebo: LS Mean | |
|---|---|---|---|---|---|
| QSYMIA
3.75 mg/23 mg | QSYMIA
15 mg/92 mg |
||||
| SD=standard deviation; SE=standard error | |||||
| Heart Rate, bpm | |||||
| Baseline mean (SD) | 73.2 (8.8) | 72.3 (9.2) | 73.1 (9.6) | +1.1 | +1.8 |
| LS Mean Change (SE) | -0.8 (0.5) | +0.3 (0.6) | +1.0 (0.5) | ||
| Systolic Blood Pressure, mmHg | |||||
| Baseline mean (SD) | 121.9 (11.5) | 122.5 (11.1) | 121.9 (11.6) | -2.8 | -3.8 |
| LS Mean Change (SE) | +0.9 (0.6) | -1.8 (0.8) | -2.9 (0.6) | ||
| Diastolic Blood Pressure, mmHg | |||||
| Baseline mean (SD) | 77.2 (7.9) | 77.8 (7.5) | 77.4 (7.7) | -0.5 | -1.9 |
| LS Mean Change (SE) | +0.4 (0.4) | -0.1 (0.6) | -1.5 (0.4) | ||
| Total Cholesterol, % | |||||
| Baseline mean (SD) | 194.3 (36.7) | 196.3 (36.5) | 192.7 (33.8) | -1.9 | -2.5 |
| LS Mean Change (SE) | -3.5 (0.6) | -5.4 (0.9) | -6.0 (0.6) | ||
| LDL-Cholesterol, % | |||||
| Baseline mean (SD) | 120.9 (32.2) | 122.8 (33.4) | 120.0 (30.1) | -2.2 | -2.8 |
| LS Mean Change (SE) | -5.5 (1.0) | -7.7 (1.3) | -8.4 (0.9) | ||
| HDL-Cholesterol, % | |||||
| Baseline mean (SD) | 49.5 (13.3) | 50.0 (11.1) | 49.7 (11.7) | +0.5 | +3.5 |
| LS Mean Change (SE) | +0.0 (0.8) | +0.5 (1.1) | +3.5 (0.8) | ||
| Triglycerides, % | |||||
| Baseline mean (SD) | 119.0 (39.3) | 117.5 (40.3) | 114.6 (37.1) | -3.9 | -14.3 |
| LS Mean Change (SE) | +9.1 (2.3) | +5.2 (3.1) | -5.2 (2.2) | ||
| Fasting Glucose, mg/dL | |||||
| Baseline mean (SD) | 93.1 (8.7) | 93.9 (9.2) | 93.0 (9.5) | -1.2 | -2.5 |
| LS Mean Change (SE) | +1.9 (0.5) | +0.8 (0.7) | -0.6 (0.5) | ||
| Waist Circumference, cm | |||||
| Baseline mean (SD) | 120.5 (14.0) | 121.5 (15.2) | 120.0 (14.7) | -2.5 † | -7.8 † |
| LS Mean Change (SE) | -3.1 (0.5) | -5.6 (0.6) | -10.9 (0.5) | ||
| Study 2 (Overweight and Obese with Comorbidities) | Placebo
(N=979) | QSYMIA
7.5 mg/46 mg (N=488) | QSYMIA
15 mg/92 mg (N=981) | QSYMIA – Placebo: LS Mean | |
|---|---|---|---|---|---|
| QSYMIA
7.5 mg/46 mg | QSYMIA
15 mg/92 mg |
||||
| SD=standard deviation; SE=standard error | |||||
| Heart Rate, bpm | |||||
| Baseline mean (SD) | 72.1 (9.9) | 72.2 (10.1) | 72.6 (10.1) | +0.6 | +1.7 |
| LS Mean Change (SE) | -0.3 (0.3) | +0.3 (0.4) | +1.4 (0.3) | ||
| Systolic Blood Pressure, mmHg | |||||
| Baseline mean (SD) | 128.9 (13.5) | 128.5 (13.6) | 127.9 (13.4) | -2.3 | -3.2 |
| LS Mean Change (SE) | -2.4 (0.48) | -4.7 (0.63) | -5.6 (0.5) | ||
| Diastolic Blood Pressure, mmHg | |||||
| Baseline mean (SD) | 81.1 (9.2) | 80.6 (8.7) | 80.2 (9.1) | -0.7 | -1.1 |
| LS Mean Change (SE) | -2.7 (0.3) | -3.4 (0.4) | -3.8 (0.3) | ||
| Total Cholesterol, % | |||||
| Baseline mean (SD) | 205.8 (41.7) | 201.0 (37.9) | 205.4 (40.4) | -1.6 | -3.0 |
| LS Mean Change (SE) | -3.3 (0.5) | -4.9 (0.7) | -6.3 (0.5) | ||
| LDL-Cholesterol, % | |||||
| Baseline mean (SD) | 124.2 (36.2) | 120.3 (33.7) | 123.9 (35.6) | +0.4 | -2.8 |
| LS Mean Change (SE) | -4.1 (0.9) | -3.7 (1.1) | -6.9 (0.9) | ||
| HDL-Cholesterol, % | |||||
| Baseline mean (SD) | 48.9 (13.8) | 48.5 (12.8) | 49.1 (13.8) | +4.0 | +5.6 |
| LS Mean Change (SE) | +1.2 (0.7) | +5.2 (0.9) | +6.8 (0.7) | ||
| Triglycerides, % | |||||
| Baseline mean (SD) | 163.5 (76.3) | 161.1 (72.2) | 161.9 (73.4) | -13.3 | -15.3 |
| LS Mean Change (SE) | +4.7 (1.7) | -8.6 (2.2) | -10.6 (1.7) | ||
| Fasting Insulin, (µIU/mL) | |||||
| Baseline mean (SD) | 17.8 (13.2) | 18.0 (12.9) | 18.4 (17.5) | -4.2 | -4.7 |
| LS Mean Change (SE) | +0.7 (0.8) | -3.5 (1.1) | -4.0 (0.8) | ||
| Fasting Glucose, mg/dL | |||||
| Baseline mean (SD) | 106.6 (23.7) | 106.2 (21.0) | 105.7 (21.4) | -2.4 | -3.6 |
| LS Mean Change (SE) | +2.3 (0.6) | -0.1 (0.8) | -1.3 (0.6) | ||
| Waist Circumference, cm | |||||
| Baseline mean (SD) | 113.4 (12.2) | 112.7 (12.4) | 113.2 (12.2) | -5.2 † | -6.8 † |
| LS Mean Change (SE) | -2.4 (0.3) | -7.6 (0.4) | -9.2 (0.3) | ||
Among the 388 subjects with type 2 diabetes treated in Study 2, reductions in HbA1c from baseline (6.8%) were 0.1% for placebo compared to 0.4% and 0.4% with QSYMIA 7.5 mg/46 mg and QSYMIA 15 mg/92 mg, respectively [see Warnings and Precautions (5.10)] .
Clinical Studies in Pediatric Patients Aged 12 Years and Older
The effect of QSYMIA on BMI in conjunction with reduced caloric intake and increased physical activity was evaluated in Study 3 (NCT 03922945), a 56-week, randomized, double-blind, placebo-controlled study in pediatric patients (12 to 17 years of age) with BMI ≥ 95 thpercentile standardized by age and sex. Patients were randomized to receive treatment with placebo (N=56), QSYMIA 7.5 mg/46 mg (N=54), or QSYMIA 15 mg/92 mg (N=113) in a 1:1:2 ratio. During the study, a well-balanced, reduced-calorie diet to result in an approximate 500 kcal/day decrease in caloric intake was recommended to all patients and patients were offered a family-based lifestyle modification program for adolescents.
Patients' mean age was 14 years old, approximately 55% were female, 67% were Caucasian, 26% were African American, and 33% were Hispanic/Latino. At the beginning of the study, the average weight and BMI of patients was 106 kg and 38 kg/m 2, respectively, with approximately 81% considered severely obese (120% of the 95 thpercentile or greater for BMI standardized by age and sex). Thirty-eight (38%) of randomized patients withdrew from the study prior to week 56.
The primary efficacy parameter was mean percent change in BMI. Table 14 provides results for BMI reduction at Week 56 in Study 3. After 56 weeks of treatment with QSYMIA, all dose levels resulted in statistically significant reduction in BMI compared to placebo (see Table 14, Figure 3). A greater proportion of patients randomized to QSYMIA than placebo achieved 5%, 10%, and 15% BMI reduction.
| Analysis Method | Placebo | QSYMIA
7.5 mg/46 mg | QSYMIA
15 mg/92 mg |
|---|---|---|---|
| SD=standard deviation; LS=least-squares; SE=standard error; CI=confidence interval | |||
|
|||
| ITT-Washout (Primary) * | n = 56 | n = 54 | n = 113 |
| BMI (kg/m 2) Primary Efficacy Endpoint | |||
| Baseline mean (SD) | 36.4 (6.4) | 36.9 (6.7) | 39.0 (7.4) |
| % LS Mean Change from baseline (SE) | +3.3 (1.4) | -4.8 (1.3) | -7.1 (1.0) |
| Difference from placebo
(95% CI) | -8.1 (-11.9, -4.3) | -10.4 (-13.9, -7.0) | |
| Percentage of patients with a reduction of greater than or equal to 5% BMI | 13.6% | 44.0% | 52.2% |
| Difference vs. placebo
(95% CI) | 29.7% (11.2, 48.3) | 38.6% (23.1, 54.1) | |
| Percentage of patients with a reduction of greater than or equal to 10% BMI | 4.5% | 33.5% | 44.4% |
| Difference vs. placebo
(95% CI) | 28.8% (13.6, 44.0) | 40.5% (28.4, 52.6) | |
| Percentage of patients with a reduction of greater than or equal to 15% BMI | 2.9% | 13.6% | 28.9% |
| Difference vs. placebo
(95% CI) | 11.7% (1.3, 22.2) | 27.4% (17.7, 37.1) | |
Figure 3. Study 3 Percent BMI Change from Baseline to Week 56 in Pediatric Patients Aged 12 to 17 Years with Obesity
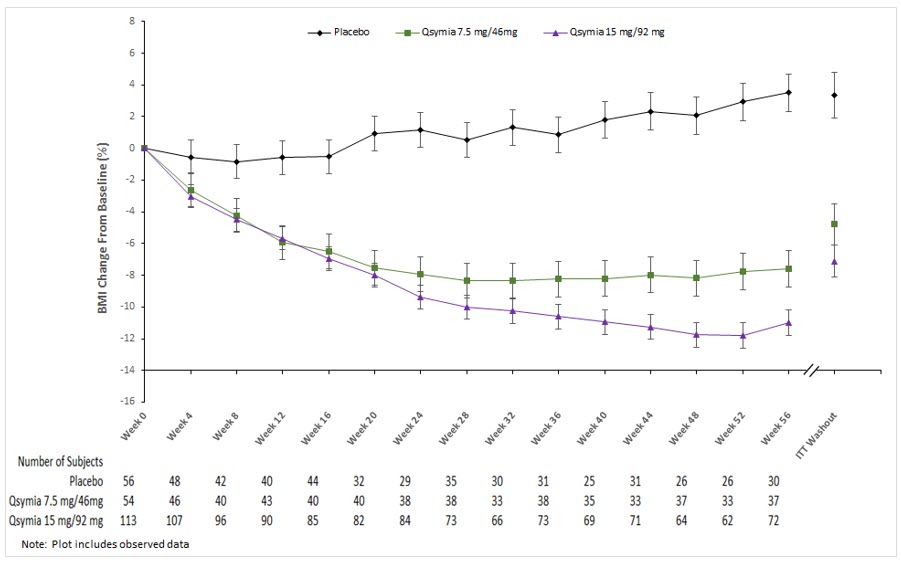
| Study 3 (Obesity)
ITT Population | Placebo
(N=56) | QSYMIA
7.5 mg/46 mg (N=54) | QSYMIA
15 mg/92 mg (N=113) | QSYMIA – Placebo | |
|---|---|---|---|---|---|
| QSYMIA
7.5 mg/46 mg | QSYMIA
15 mg/92 mg |
||||
| SD=standard deviation; SE=standard error | |||||
|
|||||
| Heart Rate, bpm * | |||||
| Baseline mean (SD) | 76.8 (9.9) | 78.6 (9.6) | 76.2 (9.6) | -5.6 | 3.2 |
| Mean Change (SD) | 2.5 (12.4) | -3.1 (8.4) | 5.7 (11.4) | ||
| Systolic Blood Pressure, mmHg | |||||
| Baseline mean (SD) | 117.7 (10.4) | 121.4 (9.2) | 117.4 (10.2) | -2.8 | -1.0 |
| LS Mean Change (SE) | +2.9 (1.6) | +0.1 (1.5) | +1.8 (1.1) | ||
| Diastolic Blood Pressure, mmHg | |||||
| Baseline mean (SD) | 71.7 (8.3) | 75.8 (6.7) | 72.9 (7.3) | -3.2 | -2.2 |
| LS Mean Change (SE) | +3.4 (1.5) | +0.2 (1.3) | +1.2 (1.0) | ||
| Total Cholesterol, mg/dL * | |||||
| Baseline mean (SD) | 164.9 (30.9) | 160.6 (26.1) | 159.4 (32.7) | -1.4 | -1.2 |
| Mean % Change (SD) | -1.8 (10.7) | -3.2 (13.1) | -3.0 (14.5) | ||
| LDL-Cholesterol, mg/dL * | |||||
| Baseline mean (SD) | 94.1 (26.8) | 89.4 (23.7) | 90.2 (27.3) | -3.9 | -2.5 |
| Mean % Change (SD) | 0.2 (23.3) | -3.7 (15.5) | -2.3 (21.6) | ||
| HDL-Cholesterol, mg/dL | |||||
| Baseline mean (SD) | 47.2 (9.7) | 47.2 (8.9) | 46.7 (10.1) | 6.4 | 5.0 |
| LS Mean % Change (SE) | -4.3 (15.1) | +2.1 (11.5) | +0.7 (9.6) | ||
| Triglycerides, mg/dL | |||||
| Baseline mean (SD) | 118.3 (46.1) | 120.1 (61.6) | 112.2 (63.2) | -11.7 | -11.2 |
| LS Mean % Change (SE) | +5.6 (8.4) | -6.2 (8.0) | -5.6 (7.2) | ||
| HbA1c, % * | |||||
| Baseline mean (SD) | 5.5 (0.3) | 5.6 (0.4) | 5.5 (0.4) | -0.2 | 0.0 |
| Mean Change (SD) | -0.2 (0.2) | -0.4 (0.3) | -0.2 (0.3) | ||
| Waist Circumference, cm | |||||
| Baseline mean (SD) | 111.1 (14.0) | 111.9 (15.5) | 116.5 (16.8) | -5.6 | -7.6 |
| LS Mean Change (SE) | +0.6 (1.4) | -5.0 (1.4) | -7.0 (1.1) | ||
16. How is Qsymia supplied
QSYMIA (phentermine and topiramate extended-release capsules) are available as follows (see Table 16):
| Strength
(phentermine mg/topiramate mg) | Description | How Supplied | NDC |
|---|---|---|---|
| 3.75 mg/23 mg extended-release capsules | Purple cap imprinted with VIVUS, Purple body imprinted with 3.75/23 | Unit of Use Bottle (14 capsules) | 62541-201-14 |
| Pharmacy Bottle (30 capsules) | 62541-201-30 | ||
| 7.5 mg/46 mg extended-release capsules | Purple cap imprinted with VIVUS, Yellow body imprinted with 7.5/46 | Unit of Use Bottle (30 capsules) | 62541-202-30 |
| 11.25 mg/69 mg extended-release capsules | Yellow cap imprinted with VIVUS, Yellow body imprinted with 11.25/69 | Pharmacy Bottle (30 capsules) | 62541-203-30 |
| 15 mg/92 mg extended-release capsules | Yellow cap imprinted with VIVUS, White body imprinted with 15/92 | Unit of Use Bottle (30 capsules) | 62541-204-30 |
| 3.75 mg/23 mg and 7.5 mg/46 mg extended-release capsules | Purple cap imprinted with VIVUS, Purple body imprinted with 3.75/23 and Purple cap imprinted with VIVUS, Yellow body imprinted with 7.5/46 | Starter Pack - Blister Configuration (28 Capsules) | 62541-210-28 |
| 11.25 mg/69 mg and 15 mg/92 mg extended-release capsules | Yellow cap imprinted with VIVUS, Yellow body imprinted with 11.25/69 | Dose Escalation Pack – Blister Configuration (28 Capsules) | 62541-220-28 |
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Embryo-Fetal Toxicity
Inform patients who can become pregnant that QSYMIA can cause fetal harm and patients should avoid getting pregnant while taking QSYMIA [see Warnings and Precautions (5.1), Drug Interactions (7), Use in Specific Populations (8.3)].
Advise patients who can become pregnant:
- that pregnancy testing is recommended before initiating QSYMIA and monthly during therapy;
- to use effective contraception during QSYMIA therapy;
- who experience spotting while taking a combined oral contraceptive to notify their healthcare provider;
- with a known or suspected pregnancy to stop QSYMIA immediately and notify their healthcare provider.
Access to QSYMIA
Advise patients that QSYMIA is only available through certified pharmacies that are enrolled in the QSYMIA certified pharmacy network. Advise patients on how to access QSYMIA through certified pharmacies. Additional information may be obtained via the website www.QSYMIAREMS.com or by telephone at 1-888-998-4887.
Increase in Heart Rate
Inform patients that QSYMIA can increase resting heart rate. Advise patients to report palpitations or feelings of a racing heartbeat while at rest to their healthcare provider(s) [see Warnings and Precautions (5.2)].
Suicidal Behavior and Ideation and Mood and Sleep Disorders
Inform patients that QSYMIA can increase the risk of mood changes, sleep disorders, depression, and suicidal ideation. Advise patients to tell their healthcare provider(s) immediately if mood changes, depression, or suicidal ideation occur [see Warnings and Precautions (5.3,5.5)].
Ophthalmologic Adverse Reactions
Inform patients that QSYMIA can increase the risk of acute myopia, secondary angle closure glaucoma, and visual field defects. Advise patients to immediately report symptoms of severe and persistent eye pain or significant changes in their vision to their healthcare provider(s) [see Warnings and Precautions (5.4)] .
Cognitive Impairment
Inform patients that QSYMIA can cause confusion, concentration, and word-finding difficulties. Inform patients that the concomitant use of alcohol or central nervous system (CNS) depressant drugs with QSYMIA, may increase the risk of dizziness, cognitive adverse reactions, drowsiness, light-headedness, impaired coordination and somnolence.
Advise patients to tell their healthcare provider(s) about any changes in attention, concentration, memory, difficulty finding words, or other cognitive functions.
Advise patients not to drive or operate machinery until they have gained sufficient experience on QSYMIA to gauge whether it adversely affects their mental performance, motor performance, and/or vision. Advise patients to avoid excessive alcohol intake while taking QSYMIA [see Warnings and Precautions (5.6)] .
Slowing of Linear Growth
Discuss with the patient and caregiver that long-term QSYMIA treatment may attenuate growth as reflected by slower height increase in pediatric patients [see Warnings and Precautions (5.7)] .
Metabolic Acidosis
Inform patients that QSYMIA can increase the risk of metabolic acidosis. Advise patients to tell their healthcare provider(s) about any factors that can increase the risk of acidosis (e.g. prolonged diarrhea, surgery, and high protein/low carbohydrate diet, and/or concomitant medications such as carbonic anhydrase inhibitors) [see Warnings and Precautions (5.8)].
Risk of Hypoglycemia in Patients with Type 2 Diabetes Mellitus on Antidiabetic Therapy
Inform patients that weight loss may increase the risk of hypoglycemia in patients with type 2 diabetes mellitus treated with insulin and/or insulin secretagogues (e.g., sulfonylureas). Advise patients with type 2 diabetes mellitus on antidiabetic therapy to monitor their blood glucose levels and report symptoms of hypoglycemia to their healthcare provider(s) [see Warnings and Precautions (5.10)] .
Risk of Hypotension in Patients Treated with Antihypertensive Medications
Advise patients that weight loss may increase the risk of hypotension. Advise patients to report symptoms of hypotension (e.g., dizziness, lightheadedness, and syncope) to the healthcare provider [see Warnings and Precautions (5.11)] .
Risk of Seizures with Abrupt Withdrawal of QSYMIA
Inform patients that abrupt withdrawal of topiramate has been associated with seizures in individuals without a history of seizures or epilepsy. Advise patients not to abruptly stop QSYMIA without first talking to their healthcare provider(s) [see Dosage and Administration (2.4), Warnings and Precautions (5.12)].
Kidney Stones
Inform patients that use of QSYMIA has been associated with kidney stone formation. Advise patients to increase fluid intake to increase urinary output which can decrease the concentration of substances involved in kidney stone formation. Advise patients to report symptoms of severe side or back pain, and/or blood in their urine to their healthcare provider(s) [see Warnings and Precautions (5.13)and Adverse Reactions (6.1)] .
Oligohidrosis and Hyperthermia
Inform patients that oligohidrosis (decreased sweating) has been reported in association with the use of topiramate particularly in pediatric patients. Advise patients to monitor for decreased sweating and increased body temperature during physical activity, especially in hot weather [see Warnings and Precautions (5.14)].
Serious Skin Reactions
Inform patients that serious skin reactions have been reported with use of topiramate. Inform patients of the signs of serious skin reactions and advise patients to report signs of a skin reaction to their healthcare provider(s) [see Warning and Precautions (5.16)] .
Lactation
Advise patients that breastfeeding is not recommended during QSYMIA treatment [see Use in Specific Populations (8.2)].
Allergic Reactions Due to Inactive Ingredient FD&C Yellow No. 5
Inform patients that this product contains FD&C Yellow No. 5 (tartrazine) which may cause allergic-type reactions (including bronchial asthma) in certain susceptible persons [see Warnings and Precautions (5.17)] .
How to Take QSYMIA
Instruct patients on the dosage titration schedule of QSYMIA. Advise patients to take QSYMIA in the morning with or without food [see Dosage and Administration (2.3)] .
Manufactured for VIVUS LLC by Catalent Pharma Solutions, LLC
1100 Enterprise Drive
Winchester, KY 40391
VIVUS LLC
900 E. Hamilton Ave., Suite 550
Campbell, CA 95008 USA
US Patent Numbers: 7,056,890; 7,553,818; 7,659,256; 7,674,776; 8,580,298; 8,580,299; 8,895,057; 8,895,058, 9,011,905; and 9,011,906
QSYMIA IS A REGISTERED TRADEMARK OF VIVUS LLC
| MEDICATION GUIDE
QSYMIA ®(Kyoo sim ee uh) (phentermine and topiramate extended-release capsules) for oral use, CIV |
|
|---|---|
| This Medication Guide has been approved by the U.S. Food and Drug Administration.
PH-03-002-14 | Revised: 06/2023 |
| What is the most important information I should know about QSYMIA?
QSYMIA can cause serious side effects, including :
QSYMIA can have other serious side effects. See " What are the possible side effects of QSYMIA?" |
|
What is QSYMIA?
|
|
Who should not take QSYMIA?Do not take QSYMIA if you:
|
|
Before taking QSYMIA, tell your healthcare provider about all of your medical conditions, including if you:
Especially tell your healthcare provider if you take:
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist each time you get a new medicine. Do notstart a new medicine without talking to your healthcare provider. |
|
How should I take QSYMIA?
|
|
What should I avoid while taking QSYMIA?
|
|
| What are the possible side effects of QSYMIA?
QSYMIA can cause serious side effects, including:
These are not all of the possible side effects of QSYMIA. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You can also report side effects to VIVUS at 1-888-998-4887. |
|
How should I store QSYMIA?
|
|
| General Information about the safe and effective use of QSYMIA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use QSYMIA for a condition for which it was not prescribed. Do not give QSYMIA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about QSYMIA that is written for health professionals. |
|
| What are the ingredients in QSYMIA?
Active Ingredient:phentermine hydrochloride and topiramate extended-release. Inactive Ingredients:FD&C Blue #1, FD&C Red #3, FD&C Yellow #5 and #6, ethylcellulose, gelatin, methylcellulose, microcrystalline cellulose, povidone, starch, sucrose, talc, titanium dioxide, and pharmaceutical black and white inks. Copyright © 2012 - 2023 VIVUS LLC All rights reserved. VIVUS LLC 900 E. Hamilton Ave., Suite 550 Campbell, CA 95008 USA US Patent Numbers: 7,056,890; 7,553,818; 7,659,256; 7,674,776; 8,580,298; 8,580,299; 8,895,057; 8,895,058; 9,011,905; and 9,011,906. QSYMIAis a registered trademark of VIVUS LLC. |
|
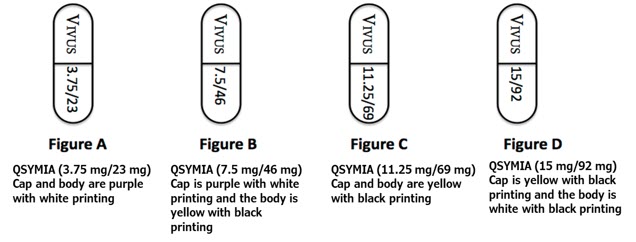
PRINCIPAL DISPLAY PANEL - 7.5 mg/46 mg Capsule Bottle Label
NDC NUMBER 62541-202-30
QSYMIA
®
(phentermine and topiramate
extended-release capsules) CIV
7.5 mg/46 mg
30 Capsules
Rx only
DISPENSE WITH MEDICATION GUIDE

PRINCIPAL DISPLAY PANEL - 3.75 mg/23 mg Capsule Bottle Label
NDC NUMBER 62541-201-30
QSYMIA
®
(phentermine and topiramate
extended-release capsules) CIV
3.75 mg/23 mg
30 Capsules
Rx only
DISPENSE WITH MEDICATION GUIDE



| QSYMIA
phentermine and topiramate capsule, extended release |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
| QSYMIA
phentermine and topiramate capsule, extended release |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| QSYMIA
phentermine and topiramate capsule, extended release |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| QSYMIA
phentermine and topiramate capsule, extended release |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - Vivus LLC (782772263) |
Frequently asked questions
More about Qsymia (phentermine / topiramate)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (564)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Support group
- FDA approval history
- Drug class: anorexiants
- En español
Patient resources
Professional resources
Related treatment guides
Copyright © 2012 - 2023 VIVUS LLC All rights reserved.