Wakix Tablets: Package Insert / Prescribing Info
Package insert / product label
Generic name: pitolisant hydrochloride
Dosage form: tablet, film coated
Drug class: Miscellaneous central nervous system agents
Medically reviewed by Drugs.com. Last updated on Jun 23, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
WAKIX® (pitolisant) tablets, for oral use
Initial U.S. Approval: 2019
Recent Major Changes
Indications and Usage for Wakix Tablets
Wakix Tablets Dosage and Administration
- See Full Prescribing Information for complete dosage instructions (2.1, 2.2)
- Administer orally once daily in the morning upon wakening (2.1, 2.2)
- Dosage Recommendations:
| Adults: EDS or Cataplexy (2.1) | |
| Week 1 | Initiate with a dosage of 8.9 mg once daily |
| Week 2 | Increase dosage to 17.8 mg once daily |
| Week 3 | May increase to the maximum recommended dosage of 35.6 mg once daily |
| Pediatric Patients (6 years and older): EDS (2.2) | |
| Week 1 | Initiate with a dosage of 4.45 mg once daily |
| Week 2 | Increase dosage to 8.9 mg once daily |
| Week 3 | Increase dosage to 17.8 mg once daily, the maximum recommended dosage for patients weighing <40 kg |
| Week 4 | For patients weighing ≥40 kg, may increase to the maximum recommended dosage of 35.6 mg once daily |
- Hepatic impairment (2.3, 8.6, 12.3):
- Moderate hepatic impairment (Child-Pugh Class B):
Adults: Initial dosage is 8.9 mg once daily. Titrate to a maximum dosage of 17.8 mg once daily after 14 days
Pediatric Patients: Initial dosage is 4.45 mg once daily. Titrate to 8.9 mg once daily after 14 days; for patients weighing ≥40 kg, may increase to 17.8 mg once daily after another 14 days
- Moderate hepatic impairment (Child-Pugh Class B):
- Renal impairment (eGFR less than 60 mL/minute/1.73 m2) (2.4, 8.7, 12.3):
- Adults: Initial dosage is 8.9 mg once daily. Titrate to a maximum dosage of 17.8 mg once daily after 7 days
- Pediatric Patients: Initial dosage is 4.45 mg once daily. Titrate to 8.9 mg once daily after 7 days; for patients weighing ≥40 kg, may increase to 17.8 mg once daily after another 7 days
- End-stage renal disease (ESRD): Not recommended
- Poor Metabolizers of CYP2D6 (2.6):
- Adults: Maximum recommended dosage is 17.8 mg once daily
- Pediatric Patients: Maximum recommended dosage is 8.9 mg once daily for patients weighing <40 kg and 17.8 mg for patients weighing ≥40 kg
Dosage Forms and Strengths
Tablets: 4.45 mg and 17.8 mg (3)
Contraindications
Warnings and Precautions
QT Interval Prolongation: Increases in QT interval. Avoid use with drugs that also increase the QT interval and in patients with risk factors for prolonged QT interval. Monitor patients with hepatic or renal impairment for increased QTc (5.1)
Adverse Reactions/Side Effects
Most common adverse reactions (≥5% and at least twice placebo) in adults: insomnia, nausea, and anxiety (6.1).
Most common adverse reactions (≥5% and greater than placebo) in pediatric patients 6 years and older: headache and insomnia (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Harmony Biosciences at 1-800-833-7460 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Strong CYP2D6 Inhibitors: Maximum recommended dosage is 17.8 mg once daily (2.5, 7.1)
- Strong CYP3A4 Inducers: Decreased exposure of WAKIX; consider dosage adjustment (2.5, 7.1)
- Sensitive CYP3A4 Substrates (including hormonal contraceptives): WAKIX may reduce effectiveness of sensitive CYP3A4 substrates. Use an alternative non-hormonal contraceptive method during treatment with WAKIX and for at least 21 days after discontinuation of treatment (7.1, 8.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2025
Full Prescribing Information
1. Indications and Usage for Wakix Tablets
WAKIX is indicated for the:
- treatment of excessive daytime sleepiness (EDS) or cataplexy in adult patients with narcolepsy.
- treatment of excessive daytime sleepiness (EDS) in pediatric patients 6 years of age and older with narcolepsy.
2. Wakix Tablets Dosage and Administration
2.1 Recommended Dosage in Adult Patients
The recommended dosage range for WAKIX for the treatment of EDS or cataplexy in adult patients is 17.8 mg to 35.6 mg administered orally once daily in the morning upon wakening.
Titrate dosage as follows:
Week 1: Initiate with a dosage of 8.9 mg (two 4.45 mg tablets) once daily
Week 2: Increase dosage to 17.8 mg (one 17.8 mg tablet) once daily
Week 3: May increase to the maximum recommended dosage of 35.6 mg (two 17.8 mg tablets) once daily
Dose may be adjusted based on tolerability.
If a dose is missed, patients should take the next dose the following day in the morning upon wakening.
It may take up to 8 weeks for some patients to achieve a clinical response.
2.2 Recommended Dosage in Pediatric Patients 6 Years and Older
The recommended starting dosage of WAKIX for the treatment of EDS in pediatric patients 6 years and older is 4.45 mg administered orally once daily in the morning upon wakening.
Titrate dosage as follows:
Week 1: Initiate with a dosage of 4.45 mg (one 4.45 mg tablet) once daily
Week 2: Increase dosage to 8.9 mg (two 4.45 mg tablets) once daily
Week 3: Increase dosage to 17.8 mg (one 17.8 mg tablet) once daily, which is the maximum recommended dosage for patients weighing <40 kg
Week 4: For patients weighing ≥40 kg, may increase to the maximum recommended dosage of 35.6 mg (two 17.8 mg tablets) once daily
Dose may be adjusted based on tolerability. If a dose is missed, patients should take the next dose the following day in the morning upon wakening. It may take up to 8 weeks for some patients to achieve a clinical response.
2.3 Dosage Recommendations in Patients with Hepatic Impairment
Adult Patients with Moderate (Child-Pugh Class B) Hepatic Impairment
- Initiate WAKIX at 8.9 mg once daily and increase after 14 days to a maximum recommended dosage of 17.8 mg once daily [see Warnings and Precautions (5.1), Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
Pediatric Patients (6 years and older) with Moderate (Child-Pugh Class B) Hepatic Impairment
- Pediatric patients weighing <40 kg: Initiate WAKIX at 4.45 mg once daily and increase after 14 days to a maximum recommended dosage of 8.9 mg once daily [see Warnings and Precautions (5.1), Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
- Pediatric patients weighing ≥40 kg: Initiate WAKIX at 4.45 mg once daily and increase after 14 days to 8.9 mg once daily. May increase after another 14 days to a maximum recommended dosage of 17.8 mg once daily [see Warnings and Precautions (5.1), Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
WAKIX is contraindicated in patients with severe hepatic impairment. WAKIX has not been studied in patients with severe hepatic impairment [see Contraindications (4), Warnings and Precautions (5.1), Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
2.4 Dosage Recommendations in Patients with Renal Impairment and End-Stage Renal Disease
Adult Patients with eGFR <60 mL/minute/1.73 m2
- Initiate WAKIX at 8.9 mg once daily and increase after 7 days to a maximum recommended dosage of 17.8 mg once daily [see Warnings and Precautions (5.1), Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
Pediatric Patients (6 years and older) with eGFR <60 mL/minute/1.73 m2 (using the Schwartz equation, eGFR (mL/min/1.73 m2)=(0.413*height in cm)/serum creatinine in mg/dL)
- Pediatric patients weighing <40 kg: Initiate WAKIX at 4.45 mg once daily and increase after 7 days to a maximum recommended dosage of 8.9 mg once daily [see Warnings and Precautions (5.1), Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
- Pediatric patients weighing ≥40 kg: Initiate WAKIX at 4.45 mg once daily and increase after 7 days to 8.9 mg once daily. May increase after another 7 days to a maximum recommended dosage of 17.8 mg once daily [see Warnings and Precautions (5.1), Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
WAKIX is not recommended in patients with eGFR less than 15 mL/minute/1.73 m2[see Warnings and Precautions (5.1), Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
2.5 Dosage Modifications for Concomitant Use with Strong CYP2D6 Inhibitors and Strong CYP3A4 Inducers
Coadministration with Strong CYP2D6 Inhibitors
- Adult patients: Initiate WAKIX at 8.9 mg once daily and increase after 7 days to a maximum recommended dosage of 17.8 mg once daily [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
- Pediatric patients (6 years and older) weighing <40 kg: Initiate WAKIX at 4.45 mg once daily and increase after 7 days to a maximum recommended dosage of 8.9 mg once daily [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
- Pediatric patients (6 years and older) weighing ≥40 kg: Initiate WAKIX at 4.45 mg once daily and increase after 7 days to 8.9 mg once daily. May increase after another 7 days to a maximum recommended dosage of 17.8 mg once daily [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
- Adult and pediatric patients on a stable dose of WAKIX: Reduce the WAKIX dose by half upon initiating strong CYP2D6 inhibitors [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Coadministration with Strong CYP3A4 Inducers
Concomitant use of WAKIX with strong CYP3A4 inducers decreases pitolisant exposure by 50%. Assess for loss of efficacy after initiation of a strong CYP3A4 inducer.
For adult and pediatric patients stable on WAKIX 8.9 mg or 17.8 mg once daily, increase the dose of WAKIX to double the original daily dose (i.e., 17.8 mg or 35.6 mg, respectively) over 7 days.
If concomitant dosing of a strong CYP3A4 inducer is discontinued, decrease WAKIX dosage by half [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.6 Dosage Recommendations in Patients Who Are Known CYP2D6 Poor Metabolizers (PMs)
Adult Patients
- Initiate WAKIX at 8.9 mg once daily and titrate to a maximum recommended dosage of 17.8 mg once daily after 7 days [see Use in Specific Populations (8.8), Clinical Pharmacology (12.5)].
Pediatric patients
- Pediatric patients weighing <40 kg: Initiate WAKIX at 4.45 mg once daily and increase after 7 days to a maximum recommended dosage of 8.9 mg once daily [see Use in Specific Populations (8.8), Clinical Pharmacology (12.5)].
- Pediatric patients weighing ≥40 kg: Initiate WAKIX at 4.45 mg once daily and increase after 7 days to 8.9 mg once daily. May increase after another 7 days to a maximum recommended dosage of 17.8 mg once daily [see Use in Specific Populations (8.8), Clinical Pharmacology (12.5)].
3. Dosage Forms and Strengths
- WAKIX 4.45 mg tablets: white, round, biconvex film-coated tablet, marked with “S” on one side and plain on the other side. Each tablet contains 5 mg of pitolisant hydrochloride equivalent to 4.45 mg of pitolisant.
- WAKIX 17.8 mg tablets: white, round, biconvex film-coated tablet, marked with “H” on one side and plain on the other side. Each tablet contains 20 mg of pitolisant hydrochloride equivalent to 17.8 mg of pitolisant.
4. Contraindications
WAKIX is contraindicated in patients with:
- known hypersensitivity to pitolisant or any component of the formulation. Anaphylaxis has been reported in patients treated with WAKIX [see Adverse Reactions (6.2)].
- severe hepatic impairment. WAKIX is extensively metabolized by the liver and there is a significant increase in WAKIX exposure in patients with moderate hepatic impairment [see Use in Specific Populations (8.6)].
5. Warnings and Precautions
5.1 QT Interval Prolongation
WAKIX prolongs the QT interval. The use of WAKIX should be avoided in patients with known QT prolongation or in combination with other drugs known to prolong the QT interval [see Drug Interactions (7.1)]. WAKIX should also be avoided in patients with a history of cardiac arrhythmias, as well as other circumstances that may increase the risk of the occurrence of torsade de pointes or sudden death, including symptomatic bradycardia, hypokalemia or hypomagnesemia, and the presence of congenital prolongation of the QT interval [see Clinical Pharmacology (12.2)]. The risk of QT prolongation may be greater in patients with hepatic or renal impairment due to higher concentrations of pitolisant. Monitor patients with hepatic or renal impairment for increased QTc. Dosage modification is recommended in patients with moderate hepatic impairment and moderate or severe renal impairment [see Dosage and Administration (2.3, 2.4)]. WAKIX is contraindicated in patients with severe hepatic impairment [see Contraindications (4)]. WAKIX is not recommended in patients with end-stage renal disease (ESRD) [see Dosage and Administration (2.4), Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in more detail in other sections of the labeling:
- QT Interval Prolongation [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Patients with Narcolepsy
In clinical trials for narcolepsy, 172 adult patients were treated with WAKIX in placebo-controlled trials for up to 8 weeks and in open-label extension trials for up to 5 years. In trials in which WAKIX was directly compared to placebo, 6 of the 152 patients (3.9%) who received WAKIX and 4 of the 114 patients (3.5%) who received placebo discontinued because of an adverse reaction.
Most Common Adverse Reactions
In the placebo-controlled clinical trials conducted in patients with narcolepsy with or without cataplexy, the most common adverse reactions (occurring in ≥5% of patients and at least twice the rate of placebo) with the use of WAKIX were insomnia (6%), nausea (6%), and anxiety (5%).
Table 1 presents the adverse reactions that occurred at a rate of ≥2% in patients treated with WAKIX and more frequently than in patients treated with placebo in the placebo-controlled clinical trials in narcolepsy.
|
* The following terms were combined: Abdominal pain includes: abdominal discomfort; abdominal pain; abdominal pain upper Anxiety includes: anxiety; nervousness; stress; stress at work Hallucinations includes: hallucination; hallucination visual; hypnagogic hallucination Headache includes: cluster headache; headache; migraine; premenstrual headache; tension headache Heart rate increased includes: heart rate increased; sinus tachycardia; tachycardia Insomnia includes: initial insomnia; insomnia; middle insomnia; poor quality sleep Musculoskeletal pain includes: arthralgia; back pain; carpal tunnel syndrome; limb discomfort; musculoskeletal pain; myalgia; neck pain; osteoarthritis; pain in extremity; sciatica Rash includes: eczema; erythema migrans; rash; urticaria Sleep disturbance includes: dyssomnia; sleep disorder; sleep paralysis; sleep talking Upper respiratory tract infection includes: pharyngitis; rhinitis; sinusitis; upper respiratory tract infection; upper respiratory tract inflammation; viral upper respiratory tract infection |
||
| Adverse Reaction | WAKIX
(n=152) % | Placebo
(n=114) % |
| Headache* | 18 | 15 |
| Insomnia* | 6 | 2 |
| Nausea | 6 | 3 |
| Upper respiratory tract infection* | 5 | 3 |
| Musculoskeletal pain* | 5 | 3 |
| Anxiety* | 5 | 1 |
| Heart rate increased* | 3 | 0 |
| Hallucinations* | 3 | 0 |
| Irritability | 3 | 2 |
| Abdominal pain* | 3 | 1 |
| Sleep disturbance* | 3 | 2 |
| Decreased appetite | 3 | 0 |
| Cataplexy | 2 | 1 |
| Dry mouth | 2 | 1 |
| Rash* | 2 | 1 |
Pediatric Patients (6 years and older) with Narcolepsy
In a clinical trial for narcolepsy, 73 pediatric patients 6 years and older were treated with WAKIX in the placebo-controlled phase for up to 8 weeks and 105 patients in the open-label extension phase for up to 6.5 years.
Most Common Adverse Reactions
In the placebo-controlled phase of the study, the most common adverse reactions (occurring in ≥5% of patients and greater than the rate of placebo) with the use of WAKIX were headache (19%) and insomnia (7%).
The overall adverse reaction profile of WAKIX in the pediatric clinical trial was similar to that seen in the adult clinical trial program.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of WAKIX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
General disorders and administration site conditions: fatigue
Immune system disorders: hypersensitivity (anaphylaxis)
Investigations: weight increased
Nervous system disorders: dizziness, epilepsy
Psychiatric disorders: abnormal behavior, abnormal dreams, anhedonia, bipolar disorder, depression, depressed mood, nightmare, sleep disorder, suicide attempt, suicidal ideation
Skin and subcutaneous tissue disorders: pruritus
Related/similar drugs
7. Drug Interactions
7.1 Drugs Having Clinically Important Interactions with WAKIX
| Effect of Other Drugs on WAKIX | |
| Strong CYP2D6 Inhibitors | |
| Clinical Implication: | Concomitant administration of WAKIX with strong CYP2D6 inhibitors increases pitolisant exposure by 2.2-fold. |
| Prevention or Management: | Reduce the dose of WAKIX by half [see see Dosage and Administration (2.5), Clinical Pharmacology (12.3)]. |
| Strong CYP3A4 Inducers | |
| Clinical Implication: | Concomitant use of WAKIX with strong CYP3A4 inducers decreases exposure of pitolisant by 50%. |
| Prevention or Management: | Assess for loss of efficacy after initiation of a strong CYP3A4 inducer. For patients stable on WAKIX 8.9 mg or 17.8 mg once daily, increase the dose of WAKIX to reach double the original daily dose (i.e., 17.8 mg or 35.6 mg, respectively) over 7 days. If concomitant dosing of a strong CYP3A4 inducer is discontinued, decrease WAKIX dosage by half [see see Dosage and Administration (2.5), Clinical Pharmacology (12.3)]. |
| Histamine-1 (H1) Receptor Antagonists | |
| Clinical Implication: | WAKIX increases the levels of histamine in the brain; therefore, H1 receptor antagonists that cross the blood-brain barrier may reduce the effectiveness of WAKIX. |
| Prevention or Management: | Avoid centrally acting H1 receptor antagonists. |
| QT Interval Prolongation | |
| Clinical Implication: | Concomitant use of drugs that prolong the QT interval may add to the QT effects of WAKIX and increase the risk of cardiac arrhythmia. |
| Prevention or Management: | Avoid the use of WAKIX in combination with other drugs known to prolong the QT interval [see Warnings and Precautions (5.1)]. |
| Effect of WAKIX on Other Drugs | |
| Sensitive CYP3A4 Substrates | |
| Clinical Implication: | WAKIX is a borderline/weak inducer of CYP3A4. Therefore, reduced effectiveness of sensitive CYP3A4 substrates may occur when used concomitantly with WAKIX [see Clinical Pharmacology (12.3)]. The effectiveness of hormonal contraceptives (e.g., ethinyl estradiol) may be reduced when used with WAKIX and effectiveness may be reduced for 21 days after discontinuation of therapy. |
| Prevention or Management: | Patients using hormonal contraception should be advised to use an alternative non-hormonal contraceptive method during treatment with WAKIX and for at least 21 days after discontinuation of treatment [see Use in Specific Populations (8.3)]. |
7.2 Drugs Having No Clinically Important Interactions with WAKIX
A clinical study was conducted to evaluate the concomitant use of WAKIX with modafinil or sodium oxybate. This study demonstrated no clinically relevant effect of modafinil or sodium oxybate on the pharmacokinetics of WAKIX and no effect of WAKIX on the pharmacokinetics of modafinil or sodium oxybate [see Clinical Pharmacology (12.3)].
A clinical study showed that strong CYP3A4 inhibitors (e.g., ketoconazole, grapefruit juice) have no effect on the pharmacokinetics of WAKIX [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women who are exposed to WAKIX during pregnancy. Patients should be encouraged to enroll in the WAKIX pregnancy registry if they become pregnant. To enroll or obtain information from the registry, patients can call 1-800-833-7460.
Risk Summary
Available case reports from clinical trials and postmarketing reports with WAKIX use in pregnant women have not determined a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproductive studies, administration of pitolisant during organogenesis caused maternal and embryofetal toxicity in rats and rabbits at doses ≥13 and >4 times the maximum recommended human dose (MRHD) of 35.6 mg based on mg/m2 body surface area, respectively. Oral administration of pitolisant to female rats during pregnancy and lactation adversely affected maternal and fetal health and produced developmental delay at doses ≥13 times the MRHD, based on mg/m2 body surface area and increased the incidence of major malformations at 22 times the MRHD (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Animal Data
Pitolisant was administered orally to pregnant rats during the period of organogenesis at doses of 30, 52, 90 and 110 mg/kg/day, which are approximately 7, 13, 22 and 27 times the MRHD, based on mg/m2 body surface area, respectively. Maternal toxicity occurred at >22 times the MRHD and included convulsions and decreases in body weight and food consumption. At these maternally toxic doses, no adverse effects on embryofetal development were noted and the no observed-adverse-effect-level for embryofetal toxicity is 27 times the MRHD based on mg/m2 body surface area.
Pitolisant was administered intramuscularly to pregnant rabbits during the period of organogenesis at doses of 4, 8, and 16 mg/kg/day, which are approximately 2, 4 and 8 times the MRHD, based on mg/m2 body surface area, respectively. Maternal toxicity occurred at ≥4 times the MRHD and included significant body weight loss and decreased food consumption. Mortality (1 animal) and convulsions (2 animals) occurred at 8 times the MRHD. At the maternally toxic dose (8 times the MRHD), the incidence of pre-implantation loss and abortions increased with a consequent decrease in both the number of implantations and live fetuses. Pitolisant was not teratogenic at doses up to 8 times the MRHD; however, delayed skeletal development (incomplete ossification and supernumerary ribs) was observed. The no-observed-adverse-effect-level for maternal toxicity and embryofetal development is 2 and 4 times the MRHD based on mg/m2 body surface area, respectively.
Pitolisant was administered orally to pregnant rats from gestation day 7 through lactation day 20 post-partum at doses of 30, 52, and 90 mg/kg/day, which are 7, 13 and 22 times the MRHD, based on mg/m2 body surface area, respectively. Maternal toxicity included death, CNS signs including convulsions, and significant decrease in body weight and food consumption at 22 times the MRHD based on mg/m2 body surface area. At the maternally toxic dose (22 times the MRHD), fetal toxicity included stillbirths, postnatal pup mortality (due to lack of milk and/or failure to nurse), and decreased pup length and weight. A single female at the mid dose (13 times the MRHD) also failed to produce milk resulting in pup mortality. At the maternally toxic dose (22 times the MRHD), pitolisant was teratogenic causing major malformations (cleft palate, abnormal limb flexure). F1 toxicity included delay in postnatal development (decrease in body weight and length, delay in incisor eruption, and delay in testes descent), which occurred at ≥13 times the MRHD; however, there was no effect on sexual maturation or reproductive capacity of the F1 generation. The no-observed-adverse-effect-level for developmental toxicity is approximately 7 times the MRHD, based on mg/m2 body surface area.
8.2 Lactation
Risk Summary
The transfer of pitolisant into breastmilk is low based on data from a lactation study. The mean infant dose was 0.009 mg/day, and the relative infant dose was less than 1% of the maternal weight-adjusted dose (see Data). There are no data on the effects of pitolisant on the breastfed infant, or the effect of this drug on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for WAKIX and any potential adverse effects on the breastfed infant from WAKIX or from the underlying maternal condition.
An open-label study in 8 healthy lactating women who were 11 to 96 weeks post-partum evaluated the concentration of pitolisant in breast milk samples collected over 24 hours and serum samples collected over 120 hours after a single dose administration of 35.6 mg of pitolisant. Pitolisant was present in breast milk with a mean Cmax of 47.5 ng/mL, while the mean Cmax of pitolisant in serum was 61.4 ng/mL. Following a single dose of pitolisant 35.6 mg, approximately 50% of the amount of pitolisant measured in breast milk occurred during the first 4 hours post dose. Based on single dose data, the mean infant dosage of pitolisant was calculated to be 0.009 mg/day, which represented a mean of 0.564% of the maternal dose received.
8.3 Females and Males of Reproductive Potential
Contraception
WAKIX may reduce the effectiveness of hormonal contraceptives. Patients using hormonal contraception should be advised to use an alternative non-hormonal contraceptive method during treatment with WAKIX and for at least 21 days after discontinuing treatment [see Drug interactions (7.1), Clinical Pharmacology (12.3)].
8.4 Pediatric Use
The safety and effectiveness of WAKIX have been established for the treatment of excessive daytime sleepiness in pediatric patients 6 years of age and older with narcolepsy. Use of WAKIX in this age group is supported by one adequate and well-controlled study in 110 pediatric patients with narcolepsy ages 6 to less than 18 years of age [see Clinical Studies (14.1)].
The safety and effectiveness of WAKIX have not been established for treatment of excessive daytime sleepiness in pediatric patients less than 6 years of age with narcolepsy.
The safety and effectiveness of WAKIX have not been established for treatment of cataplexy in pediatric patients with narcolepsy.
Juvenile Animal Toxicity Data
In a juvenile animal study, male and female rats were administered pitolisant at 9, 21, or 48 mg/kg/day by oral gavage from postnatal day (PND) 7 to PND 70. Mortality occurred at the highest dose of 48 mg/kg/day; however, death was primarily related to aspiration/inhalation of food material. No adverse effects on growth and development up to the high dose were observed; however, plasma exposures at this dose were lower than those predicted to occur in pediatric patients at the maximum recommended human dose (MRHD) of 35.6 mg due to low oral bioavailability in juvenile rats.
In a second juvenile animal study, male and female rats were administered pitolisant at 15 or 30 mg/kg/day or 30 mg/kg/twice daily (60 mg/kg/day) by intraperitoneal injection from PND 7 to PND 70. Mortality and convulsions were observed at the top two doses of 30 and 60 mg/kg/day. Similar findings of convulsions and mortality were also observed in studies in adult rats at comparable doses. The no-observed-adverse-effect-level (NOAEL) is 15 mg/kg/day in juvenile animals administered pitolisant by intraperitoneal injection, which corresponds to plasma exposures that are approximately 4 times and 1 times the predicted pediatric exposures at the MRHD of 35.6 mg, based on Cmax and AUC, respectively.
8.5 Geriatric Use
Limited pharmacokinetic data are available in healthy elderly subjects. A pharmacokinetic study that compared 12 elderly subjects (age 68 to 82 years) to 12 healthy adults (age 18 to 45 years) did not reveal any significant differences in drug exposure [see Clinical Pharmacology (12.3)].
Of the total number of patients with narcolepsy in clinical studies of WAKIX, 14 patients (5%) were ≥65 years old. No overall differences in safety or effectiveness were observed between these patients and younger patients in these clinical trials, but greater sensitivity of some older individuals cannot be ruled out. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function, concomitant diseases, and other drug therapy.
8.6 Hepatic Impairment
WAKIX is contraindicated in patients with severe hepatic impairment (Child-Pugh Class C) as it has not been studied in this population. WAKIX is extensively metabolized by the liver and there is a significant increase in WAKIX exposure in patients with moderate hepatic impairment [see Contraindications (4), Clinical Pharmacology (12.3)].
Monitor patients with moderate hepatic impairment (Child-Pugh Class B) and adjust the dosage of WAKIX [see Dosage and Administration (2.3)].
Monitor patients with mild hepatic impairment (Child-Pugh Class A). No dosage adjustment of WAKIX is recommended in patients with mild hepatic impairment.
8.7 Renal Impairment
The pharmacokinetics of WAKIX in patients with end-stage renal disease (ESRD) (eGFR of <15 mL/minute/1.73 m2) is unknown [see Clinical Pharmacology (12.3)]. Therefore, WAKIX is not recommended in patients with ESRD [see Dosage and Administration (2.4), Warnings and Precautions (5.1)].
Dosage adjustment of WAKIX is recommended in patients with eGFR <60 mL/minute/1.73 m2[see Dosage and Administration (2.4)].
10 OVERDOSAGE
Human Experience
In premarketing clinical studies, overdosage of WAKIX was identified in one patient who ingested 320.4 mg of WAKIX (9 times the maximum recommended daily dose). The patient experienced nystagmus and twitching, reported generalized pain, and had QT prolongation. The patient was monitored in the hospital, received supportive care, and was discharged the following day without sequelae.
There have been postmarketing reports of overdoses of WAKIX. The most frequently reported symptoms with overdoses up to 71.2 mg (twice the maximum recommended daily dose) were headache, nausea, and hallucinations.
Management of Overdosage
No specific antidote for WAKIX is known. In managing acute overdosage, provide general symptomatic and supportive care including close medical supervision and monitoring of vital signs and cardiac rhythm. Specifically, continuous cardiovascular monitoring should be initiated to detect possible arrhythmias [see Warnings and Precautions (5.1)]. Consider contacting the Poison Help line (1-800-222-1222) or a medical toxicologist for additional overdose management recommendations.
11. Wakix Tablets Description
WAKIX tablets contain pitolisant hydrochloride. Pitolisant is an antagonist/inverse agonist of the histamine-3 (H3) receptor. Pitolisant hydrochloride is a white or almost white crystalline powder with a molecular formula of C17H26ClNO·HCl and a molecular weight of 332.31. Pitolisant hydrochloride is soluble in water, ethanol, and methylene chloride and practically insoluble in cyclohexane. The chemical name of pitolisant hydrochloride is 1-{3-[3-(4-chlorophenyl)propoxy]propyl}piperidine, hydrochloride and its structural formula is:
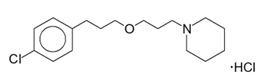
The molecular formula of the pitolisant free base is C17H26ClNO and its molecular weight is 295.85.
WAKIX tablets are for oral administration and each film-coated tablet contains 5 mg or 20 mg of pitolisant hydrochloride (equivalent to 4.45 mg or 17.8 mg of pitolisant free base, respectively) and the following inactive ingredients: colloidal silicon dioxide, crospovidone, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
12. Wakix Tablets - Clinical Pharmacology
12.1 Mechanism of Action
The mechanism of action of pitolisant in excessive daytime sleepiness (EDS) in patients 6 years and older with narcolepsy or cataplexy in adult patients with narcolepsy is unclear. However, its efficacy could be mediated through its activity as an antagonist/inverse agonist at histamine-3 (H3) receptors.
12.2 Pharmacodynamics
Pitolisant binds to H3 receptors with a high affinity (Ki = 1 nM) and has no appreciable binding to other histamine receptors (H1, H2, or H4 receptors; Ki ≥10 µM).
Cardiac Electrophysiology
WAKIX at the highest recommended dosage (i.e., 35.6 mg daily) led to a QTc increase of 4.2 msec. Exposures 3.8-fold higher than achieved at the highest recommended dose increased QTc 16 msec (mean) [see Warnings and Precautions (5.1)].
12.3 Pharmacokinetics
Following oral administration of pitolisant 35.6 mg once daily, the steady state Cmax and AUC are 73 ng/mL (range: 49.2 to 126 ng/mL) and 812 ng*hr/mL (range: 518 to 1468 ng*hr/mL), respectively. Pitolisant exposure (Cmax and AUC) increases proportionally with dose and steady state is reached by day 7.
Absorption
The median time to maximum plasma concentration (Tmax) of pitolisant is 3.5 hours (2 to 5 hours).
The oral absorption of WAKIX is around 90%.
Food Effect
No clinically significant differences in the pharmacokinetics of pitolisant were observed following administration with a high-fat meal.
Distribution
The apparent volume of distribution of pitolisant is approximately 700 L (5 to 10 L/kg). Serum protein binding is approximately 91% to 96%. The blood to plasma ratio of pitolisant is 0.55 to 0.89.
Elimination
After a single dose of 35.6 mg, the median half-life of pitolisant is approximately 20 hours (7.5 to 24.2 hours). The apparent oral clearance (CL/F) of pitolisant is 43.9 L/hr and renal clearance accounts for <2% of the total clearance of pitolisant.
Metabolism
Pitolisant is primarily metabolized by CYP2D6 and to a lesser extent by CYP3A4; these metabolites are further metabolized or conjugated with glycine or glucuronic acid. None of these metabolites are pharmacologically active.
Excretion
After a single oral radiolabeled pitolisant 17.8 mg dose, approximately 90% of the dose was excreted in urine (<2% unchanged) and 2.3% in feces.
Specific Populations
No clinically significant differences in the pharmacokinetics of pitolisant were observed based on age (18 to 82 years old), sex, race/ethnicity (Caucasians or Blacks), or body weight (48 to 103 kg). The effects of end-stage renal disease and severe hepatic impairment on the pharmacokinetics of pitolisant are unknown.
Pediatric Patients
Pharmacokinetic data from 24 pediatric patients with narcolepsy (ages 7 to 17 years) receiving a single dose of WAKIX suggest that pediatric patients have higher exposure to pitolisant than adults. The geometric mean Cmax and AUC0-10h of pitolisant were 2.2 and 2.0-fold higher, respectively, in pediatric patients 12 to 17 years and 3.4 and 3.6-fold higher, respectively, in pediatric patients 7 to 11 years compared to adults.
Patients with Hepatic Impairment
Six subjects with mild hepatic impairment (Child-Pugh Class A), 6 subjects with moderate hepatic impairment (Child-Pugh Class B), and 12 healthy subjects matched for age, sex, body mass index and ethnicity received a single dose of WAKIX 17.8 mg to assess the pharmacokinetics of WAKIX in patients with hepatic impairment. Exposure of pitolisant in patients with mild or moderate hepatic impairment is summarized in Figure 1. No studies have been conducted in patients with severe hepatic impairment.
Figure 1: Effect of Hepatic Impairment on Pitolisant Pharmacokinetics
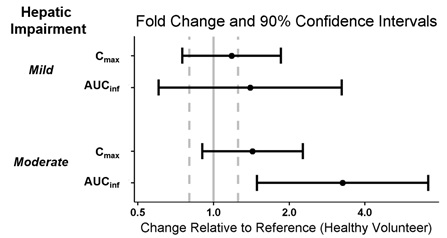
Dots = Geometric least squares mean ratios, Error bars = 90% CI; reference dashed lines are 0.8 and 1.25.
AUCinf = area under the curve from time 0 to time infinity; Cmax = maximum plasma concentration.
Patients with Renal Impairment
A single dose of WAKIX 17.8 mg was administered to 4 subjects with mild renal impairment (eGFR of 60 to 89 mL/min/1.73 m2), 4 subjects with moderate renal impairment (eGFR of 30 to 59 mL/min/1.73 m2), 4 subjects with severe renal impairment (eGFR of 15 to 29 mL/min/1.73 m2), and 12 subjects with normal renal function (i.e., eGFR >90 mL/min/1.73 m2) to assess the pharmacokinetics of WAKIX in patients with renal impairment. Exposure of pitolisant in patients with mild, moderate, and severe renal impairment is summarized in Figure 2. No studies have been conducted in patients with ESRD.
Figure 2: Effect of Renal Impairment on Pitolisant Pharmacokinetics
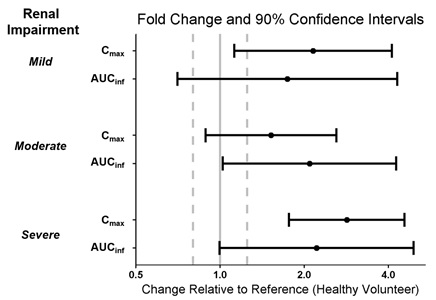
Dots = Geometric least squares mean ratios, Error bars = 90% CI; reference dashed lines are 0.8 and 1.25.
AUCinf = area under the curve from time 0 to time infinity; Cmax = maximum plasma concentration.
CYP2D6 Poor Metabolizers
The pharmacokinetics of pitolisant were evaluated in 3 subjects who were CYP2D6 poor metabolizers (PMs) and 5 subjects who were CYP2D6 extensive metabolizers (EMs). All subjects received WAKIX 17.8 mg daily for 7 days. Exposure of pitolisant in CYP2D6 PMs is summarized in Figure 3.
Figure 3: Pitolisant Pharmacokinetics in CYP2D6 Poor Metabolizers
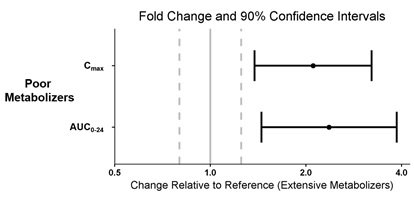
Dots = Geometric least squares mean ratios, Error bars = 90% CI; reference dashed lines are 0.8 and 1.25.
AUC0-24 = area under the curve from time 0 to 24 hours post-dose; Cmax = maximum plasma concentration.
Drug-Drug Interactions
Effect of Other Drugs on the Pharmacokinetics of WAKIX
The effect of other drugs on the pharmacokinetics of pitolisant is presented in Figure 4 [see Dosage and Administration (2.5), Drug Interactions (7.1)].
Figure 4: Effect of Concomitant Medications on Pitolisant
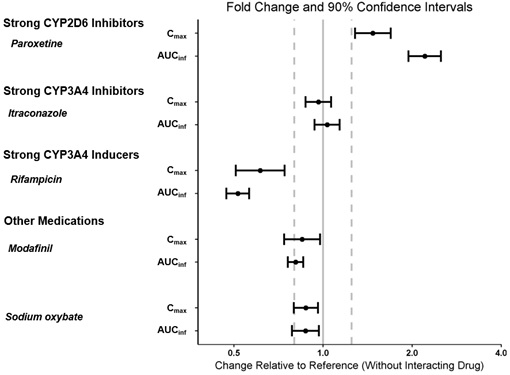
Dots = Geometric least squares mean ratios, Error bars = 90% CI; reference dashed lines are 0.8 and 1.25.
AUCinf = area under the curve from time 0 to time infinity; Cmax = maximum plasma concentration.
Effect of WAKIX on the Pharmacokinetics of Other Drugs
The effect of pitolisant on the pharmacokinetics of other drugs is presented in Figure 5[see Drug Interactions (7.1, Use in Specific Populations (8.3)].
Figure 5: Effect of Pitolisant on Concomitant Medications
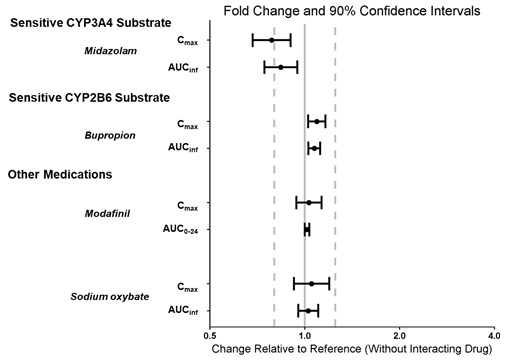
Dots = Geometric least squares mean ratios, Error bars = 90% CI; reference dashed lines are 0.8 and 1.25.
AUCinf = area under the curve from time 0 to time infinity; AUC0-24 = area under the curve from time 0 to 24 hours; Cmax = maximum plasma concentration.
12.5 Pharmacogenomics
Approximately 3 to 10% of Caucasians and 2 to 7% of African Americans generally lack the capacity to metabolize CYP2D6 substrates and are classified as poor metabolizers. The AUC of pitolisant was approximately 2.4 times higher in CYP2D6 poor metabolizers than in normal metabolizers and is similar to the exposure of pitolisant when WAKIX is administered concomitantly with a CYP2D6 inhibitor [see Dosage and Administration (2.6), Drug Interactions (7.1)].
In CYP2D6 poor metabolizers, the Cmax of pitolisant is 153 (151 to 157) ng/mL and the AUC is 1920 (1854 to 2000) ng*hr/mL after steady state dosing with 35.6 mg once daily.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Pitolisant was not carcinogenic in mice or rats.
Oral administration of pitolisant at 15, 30, and 75 mg/kg/day for 6 months to CB6F1 TgrasH2 transgenic mice did not increase tumor incidence. These doses are 2, 4, and 9 times the MRHD, respectively, based on mg/m2 body surface area.
Oral administration of pitolisant at 5, 15, and 30 mg/kg/day for 105 weeks to Sprague-Dawley rats did not increase tumor incidence. These doses are 1.4, 4, and 8 times the MRHD based on mg/m2 body surface area.
Mutagenesis
Pitolisant and its metabolites were not mutagenic in the in vitro bacterial reverse mutation assay (Ames), or clastogenic in the in vitro mammalian chromosomal aberration assay. Pitolisant was negative in the in vivo mouse micronucleus assay.
Impairment of Fertility
Oral administration of pitolisant at 30, 52, and 90 mg/kg/day to male and female rats prior to and throughout mating and continuing in females through early gestation resulted in adverse effects at the mid and high doses. These doses are 13 and 22 times the MRHD, respectively, based on mg/m2 body surface area (BSA). A dose-related increase in the percentage of post-implantation loss was observed compared to controls, leading to a decrease in the percentage of live conceptuses, at doses 13 and 22 times the MRHD, based on mg/m2 BSA. Pitolisant caused dose-related abnormalities in sperm morphology and decreased motility at doses that are 13 and 22 times the MRHD based on mg/m2 BSA, without any significant effects on fertility indices in male rats. No effects on fertility were observed at 30 mg/kg/day (7 times the MRHD based on mg/m2 BSA).
13.2 Animal Toxicology and/or Pharmacology
Adverse CNS-related clinical signs including tremors and convulsions occurred after single and repeated oral administration of pitolisant across multiple species. In a 9-month repeat-dose toxicity study in adult monkeys, sporadic incidences of convulsions occurred at doses corresponding to exposures approximately 3 times the MRHD based on Cmax and 1 times the MRHD, based on AUC. Convulsions were first observed close to Tmax and resolved by 2 hours after dosing. Convulsions were not observed after discontinuation of dosing and were not associated with microscopic findings in the brain. Safety margins at the no-observed-adverse-effect-level (NOAEL) correspond to 1 times the MRHD based on Cmax and 0.4 times based on AUC.
14. Clinical Studies
14.1 Excessive Daytime Sleepiness (EDS) in Patients with Narcolepsy
Adult Patients with Narcolepsy
The efficacy of WAKIX for the treatment of excessive daytime sleepiness in adult patients with narcolepsy was evaluated in two multicenter, randomized, double-blind, placebo-controlled studies (Study 1; NCT01067222 and Study 2; NCT01638403). Patients ≥18 years of age who met the International Classification of Sleep Disorders (ICSD-2) criteria for narcolepsy (with or without cataplexy) and who had an Epworth Sleepiness Scale (ESS) score ≥14 were eligible to enroll in the studies. EDS was assessed using the ESS, an 8-item questionnaire by which patients rate their perceived likelihood of falling asleep during usual daily life activities. Each of the 8 items on the ESS is rated from 0 (would never doze) to 3 (high chance of dozing); the maximum score is 24. Study 1 and Study 2 included an 8-week treatment period: a 3-week dose titration phase followed by a 5-week stable dose phase. These studies compared WAKIX to both a placebo and an active control.
In Study 1, 95 patients were randomized to receive WAKIX, placebo, or active control. The dose of WAKIX was initiated at 8.9 mg once daily and could be increased at weekly intervals to 17.8 mg or 35.6 mg, based on clinical response and tolerability. No dose adjustments were permitted during the 5-week stable dose phase. 61% of patients reached a stable dose of 35.6 mg. Median age in the study was 37 years. More than 90% of patients in the WAKIX and placebo groups were Caucasian and 54% were male. Approximately 80% of the population had a history of cataplexy.
WAKIX demonstrated statistically significantly greater improvement on the primary endpoint, the least squares mean final ESS score compared to placebo (Table 3).
In Study 2, 166 patients were randomized to receive WAKIX, placebo, or active control. The dose of WAKIX was initiated at 4.45 mg and could be increased at weekly intervals to 8.9 mg or 17.8 mg, based on clinical response and tolerability. No dose adjustments were permitted during the 5-week stable-dose phase. 76% of patients reached a stable dose of 17.8 mg. Median age in the study was 40 years. In the WAKIX and placebo groups, approximately 50% of patients were male, 90% of patients were Caucasian, and 75% of patients had a history of cataplexy.
WAKIX demonstrated statistically significantly greater improvement on the primary endpoint, the least squares mean final ESS score compared to placebo (Table 3).
Examination of demographic subgroups by sex did not suggest differences in response.
The efficacy results from Study 1 and Study 2 are shown in Table 3.
|
CI = confidence interval; LS Mean = least squares mean; SD = standard deviation; SE = standard error aMaximum dose randomized to was 35.6 mg bMaximum dose randomized to was 17.8 mg cA lower score on the ESS represents improvement; scores range from 0 (no symptoms) to 24 (worst symptoms) dA negative value for the placebo subtracted difference represents improvement |
||||
| Study | Treatment
Group (N) | Baseline ESS Score
Mean (SD) | Final ESS Scorec
LS Mean at Week 8 (SE) | Placebo Subtracted
Difference [95% CI] at Week 8d |
| Study 1a | WAKIX (n=31) | 17.8 (2.5) | 12.4 (1.01) | -3.1 [-5.73; -0.46] |
| Placebo (n=30) | 18.9 (2.5) | 15.5 (1.03) | ||
| Study 2b | WAKIX (n=66) | 18.3 (2.4) | 13.3 (1.19) | -2.2 [-4.17; -0.22] |
| Placebo (n=32) | 18.2 (2.3) | 15.5 (1.32) | ||
Figure 6 shows the ESS score from baseline to Week 8 in Study 1.
Figure 6: Epworth Sleepiness Scale Score (mean ± SEM) from Baseline to Week 8 in Study 1
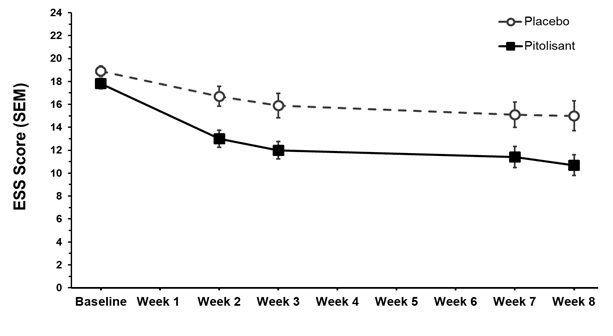
SEM = standard error of the mean (raw mean scores)
ESS scores range from 0 to 24, with 0 being the best score and 24 being the worst score
Pediatric Patients (6 years of age and older) with Narcolepsy
The efficacy of WAKIX for the treatment of excessive daytime sleepiness in pediatric patients 6 years of age and older with narcolepsy was evaluated in one multicenter, randomized, double-blind, placebo-controlled study (Study 4; NCT02611687). Pediatric patients 6 to 17 years who met the International Classification of Sleep Disorders (ICSD-3) criteria for narcolepsy (with or without cataplexy) and who had a Pediatric Daytime Sleepiness Scale (PDSS) score ≥15 were eligible to enroll in the study. EDS was assessed with the PDSS, an 8-item questionnaire in which patients report their frequency of EDS-related symptoms. Each of the 8 items on the PDSS is rated from 0 (never) to 4 (very often, always); the maximum score is 32, with higher scores representing greater severity of symptoms. The study included an 8-week treatment period: a 4-week dose titration phase followed by a 4-week stable dose phase.
In Study 4, 110 pediatric patients 6 to 17 years were randomized to receive WAKIX or placebo. The dose of WAKIX was initiated at 4.45 mg once daily and could be increased at weekly intervals to 17.8 mg for patients weighing <40 kg or 35.6 mg for patients weighing≥40 kg, based on clinical response and tolerability. No dose adjustments were permitted during the 4-week stable dose phase. 69% of patients weighing <40 kg reached a stable dose of 17.8 mg and 72% of patients weighing ≥40 kg reached a stable dose of 35.6 mg. Median age in the study was 13 years, 56% of the patients were male, and 82% of the population had a history of cataplexy. Race and ethnicity were not collected in this study.
WAKIX demonstrated statistically significantly greater improvement on the least squares mean change from baseline to the end of treatment in final PDSS total score compared to placebo, of -3.41 points (95% CI: -5.52, -1.31). Study 4 included global assessments, which showed positive trends supporting PDSS total score of improvement in favor of WAKIX.
14.2 Cataplexy in Adult Patients with Narcolepsy
The efficacy of WAKIX for the treatment of cataplexy in adult patients with narcolepsy was evaluated in two multicenter, randomized, double-blind, placebo-controlled studies (Study 3; NCT01800045 and Study 1; NCT01067222). Patients ≥18 years of age who met the International Classification of Sleep Disorders (ICSD-2) criteria for narcolepsy with cataplexy with at least 3 cataplexy attacks per week and an ESS score of ≥12 were eligible to enroll in Study 3; patients meeting the ICSD-2 criteria for narcolepsy (with or without cataplexy) and an ESS score of ≥14 were eligible to enroll in Study 1.
Study 3 included a 7-week treatment period: a 3-week dose titration phase followed by a 4-week stable dose phase. 105 patients were randomized to receive WAKIX or placebo. The dose of WAKIX was initiated at 4.45 mg once daily for the first week, increased to 8.9 mg for the second week, and could remain the same or be decreased or increased at the next two weekly intervals to a maximum of 35.6 mg, based on clinical response and tolerability. No dose adjustments were permitted during the 4-week stable dose phase. 65% of patients reached a stable dose of 35.6 mg. Median age in the study was 37 years and 51% of the patients were male. Race was not collected in this study.
WAKIX demonstrated statistically significantly greater improvement on the primary endpoint, the change in geometric mean number of cataplexy attacks per week from baseline to the average of the 4 week stable dosing period for WAKIX compared to placebo (Table 4).
Study 1 was described in Section 14.1. In the subset of patients with a history of cataplexy (n=49), WAKIX demonstrated statistically significantly greater improvement on the secondary endpoint, the change from baseline in geometric mean daily rate of cataplexy at Week 8 for WAKIX compared to placebo (Table 4).
In both Study 3 and Study 1, examination of demographic subgroups by sex did not suggest differences in response.
|
CI = confidence interval; SD = standard deviation a The mean refers to geometric mean, which was used because values for weekly rate of cataplexy were not normally distributed; the geometric mean takes the average of the logs of the individual values and exponentiates that average back to an arithmetic scale. b The rate ratio is derived from a Poisson regression model. c Patients with a history of cataplexy. |
|||||
| Study | Endpoint | Treatment Group (N) | Baseline Rate Meana (SD) [95% CI] | Final Rate Meana (SD) [95% CI] | Rate Ratiob
(WAKIX to placebo) [95% CI] |
|---|---|---|---|---|---|
| Study 3 | Final Mean Weekly Rate of Cataplexy Over 4-Week Stable Dosing Period (primary endpoint) | WAKIX (n=54) | 9.1 (2.0) [7.6, 11.0] | 2.3 (4.4) [1.5, 3.4] | 0.51 [0.44, 0.60] |
| Placebo (n=51) | 7.3 (2.0) [6.0, 8.9] | 4.5 (4.8) [2.9, 7.0] |
|||
| Study 1 | Final Mean Daily Rate of Cataplexy at Week 8 (secondary endpoint) | WAKIX (n=25)c | 0.4 (4.0) [0.2, 0.7] | 0.1 (2.8) [0.1, 0.2] | 0.07 [0.01, 0.36] |
| Placebo (n=24) c | 0.3 (3.6) [0.1, 0.4] | 0.2 (4.3) [0.1, 0.5] |
|||
16. How is Wakix Tablets supplied
16.1 How Supplied
WAKIX (pitolisant) tablets are available as:
4.45 mg: white, round, biconvex film-coated tablet, 3.7 mm diameter, marked with “S” on one side and plain on the other side.
NDC 72028-045-03 – Bottles of 30
17.8 mg: white, round, biconvex film-coated tablet, 7.5 mm diameter, marked with “H” on one side and plain on the other side.
NDC 72028-178-03 – Bottles of 30
17. Patient Counseling Information
Prolongation of the QT Interval
Inform patients to consult their physician immediately if they feel faint, lose consciousness, or have heart palpitations [see Warnings and Precautions (5.1)]. Advise patients to inform their healthcare provider that they are taking WAKIX before any new drug is taken.
Contraception
Advise patients that use of WAKIX may reduce the efficacy of hormonal contraceptives. Advise patients using a hormonal contraceptive to use an alternative non-hormonal contraceptive method of contraception during treatment and for at least 21 days after discontinuing treatment [see Drug Interactions (7.1), Use in Specific Populations (8.3)].
Pregnancy
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to WAKIX during pregnancy [see Use in Specific Populations (8.1)].
Concomitant Medication
Advise patients to inform their healthcare provider if they are taking, or plan to take, any prescription or over-the-counter drugs, because of the potential for interactions between WAKIX and other drugs [see Drug Interactions (7)].
Distributed by: Harmony Biosciences, LLC, Plymouth Meeting, PA 19462 USA
WAKIX is a registered trademark of Bioprojet Europe, Ltd.
Protected by US Patent Numbers 8,207,197; 8,354,430; 8,486,947
Harmony Biosciences name and logo are trademarks of Harmony Biosciences Management, Inc. and are used herein by permission.
Label #100.09



| WAKIX
pitolisant hydrochloride tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| WAKIX
pitolisant hydrochloride tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Harmony Biosciences, LLC (080960093) |
| Registrant - Harmony Biosciences Management Inc. (178031683) |
Frequently asked questions
More about Wakix (pitolisant)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (22)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: miscellaneous central nervous system agents
- Breastfeeding
- En español
