Zurzuvae: Package Insert / Prescribing Info
Package insert / product label
Dosage form: capsule
Drug class: Miscellaneous antidepressants
Medically reviewed by Drugs.com. Last updated on Nov 25, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Drug Abuse and Dependence
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
ZURZUVAETM (zuranolone) capsules, for oral use, CIV
Initial U.S. Approval: 2023
WARNING: IMPAIRED ABILITY TO DRIVE OR ENGAGE IN OTHER POTENTIALLY HAZARDOUS ACTIVITIES
See full prescribing information for complete boxed warning.
ZURZUVAE causes driving impairment due to central nervous system (CNS) depressant effects. Advise patients not to drive or engage in other potentially hazardous activities until at least 12 hours after administration. Patients may not be able to assess their own driving competence or the degree of impairment caused by ZURZUVAE (5.1, 5.2).
Indications and Usage for Zurzuvae
ZURZUVAE is a neuroactive steroid gamma-aminobutyric acid (GABA) A receptor positive modulator indicated for the treatment of postpartum depression (PPD) in adults. (1)
Zurzuvae Dosage and Administration
- Administer with fat-containing food. (2.1)
- Recommended dosage is 50 mg orally once daily in the evening for 14 days. (2.1)
- Dosage may be reduced to 40 mg once daily if CNS depressant effects occur. (2.1)
- ZURZUVAE can be used alone or as an adjunct to oral antidepressant therapy. (2.1)
- Severe Hepatic Impairment: Recommended dosage is 30 mg orally once daily in the evening for 14 days. (2.3, 8.6)
- Moderate or Severe Renal Impairment: Recommended dosage is 30 mg orally once daily in the evening for 14 days. (2.4, 8.7)
Dosage Forms and Strengths
Capsules: 20 mg, 25 mg, and 30 mg. (3)
Contraindications
None. (4)
Warnings and Precautions
- CNS Depressant Effects: ZURZUVAE can cause CNS depressant effects such as somnolence and confusion. If patients develop CNS depression, consider dosage reduction or discontinuation of ZURZUVAE. (5.2)
- Suicidal Thoughts and Behavior: Consider changing the therapeutic regimen, including discontinuing ZURZUVAE, in patients whose PPD worsens, or who experience emergent suicidal thoughts and behaviors. (5.3)
- Embryo-fetal Toxicity: May cause fetal harm. Advise a pregnant woman of the potential risk to an infant exposed to ZURZUVAE in utero. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during ZURZUVAE treatment and for one week after the final dose. (5.4, 8.1, 8.2, 8.3)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥5% and greater than placebo) were somnolence, dizziness, diarrhea, fatigue, nasopharyngitis, and urinary tract infection. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Biogen at 1-844-987-9882 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- CNS Depressants: Concomitant use may increase impairment of psychomotor performance or CNS depressant effects. If use with another CNS depressant is unavoidable, consider dosage reduction. (7)
- Strong CYP3A4 Inhibitors: Concomitant use may increase the risk of ZURZUVAE-associated adverse reactions. Reduce the ZURZUVAE dosage to 30 mg orally once daily in the evening for 14 days when used concomitantly with a strong CYP3A4 inhibitor. (2.2, 7)
- CYP3A4 Inducers: Concomitant use may decrease the efficacy of ZURZUVAE. Avoid concomitant use. (2.2, 7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2024
Full Prescribing Information
WARNING: IMPAIRED ABILITY TO DRIVE OR ENGAGE IN OTHER POTENTIALLY HAZARDOUS ACTIVITIES
ZURZUVAE causes driving impairment due to central nervous system (CNS) depressant effects [see Warnings and Precautions (5.1, 5.2)].
Advise patients not to drive or engage in other potentially hazardous activities until at least 12 hours after ZURZUVAE administration for the duration of the 14-day treatment course. Inform patients that they may not be able to assess their own driving competence, or the degree of driving impairment caused by ZURZUVAE [see Warnings and Precautions (5.1)].
1. Indications and Usage for Zurzuvae
ZURZUVAE is indicated for the treatment of postpartum depression (PPD) in adults.
2. Zurzuvae Dosage and Administration
2.1 Recommended Dosage
The recommended dosage of ZURZUVAE is 50 mg taken orally once daily in the evening for 14 days. Administer ZURZUVAE with fat-containing food (e.g., 400 to 1,000 calories, 25% to 50% fat) [see Clinical Pharmacology (12.3)]. If patients experience CNS depressant effects within the 14-day period, consider reducing the dosage to 40 mg once daily in the evening within the 14-day period.
ZURZUVAE can be used alone or as an adjunct to oral antidepressant therapy.
The safety and effectiveness of ZURZUVAE use beyond 14 days in a single treatment course have not been established.
2.2 Dosage Modifications for Concomitant Use with CYP3A4 Inducers or CYP3A4 Inhibitors
CYP3A4 Inducers
Avoid concomitant use of ZURZUVAE with CYP3A4 inducers [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
CYP3A4 Inhibitors
Reduce the ZURZUVAE dosage to 30 mg orally once daily in the evening for 14 days when used concomitantly with a strong CYP3A4 inhibitor [see Drug Interactions (7) and Clinical Pharmacology (12.3)]. No dosage modification is recommended when ZURZUVAE is concomitantly used with a moderate CYP3A4 inhibitor.
2.3 Recommended Dosage in Patients with Hepatic Impairment
The recommended dosage of ZURZUVAE in patients with severe hepatic impairment (Child-Pugh C) is 30 mg orally once daily in the evening for 14 days [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)]. The recommended dosage in patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment is the same as those in patients with normal hepatic function.
2.4 Recommended Dosage in Patients with Renal Impairment
The recommended dosage of ZURZUVAE in patients with moderate or severe renal impairment (eGFR <60 mL/min/1.73 m2) is 30 mg orally once daily in the evening for 14 days [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
The recommended dosage in patients with mild renal impairment (eGFR 60 to 89 mL/min/1.73 m2) is the same as those in patients with normal renal function.
2.5 Recommendations Regarding a Missed Dose
If a ZURZUVAE evening dose is missed, take the next dose at the regular time the following evening. Do not take extra capsules on the same day to make up for the missed dose. Continue taking ZURZUVAE once daily until the remainder of the 14-day treatment course is completed.
3. Dosage Forms and Strengths
Capsules:
- 20 mg: light-orange cap, ivory to light-yellow body, printed with “S-217 20 mg” on body of capsule in black
- 25 mg: light-orange cap, light-orange body, printed with “S-217 25 mg” on body of capsule in black
- 30 mg: orange cap, light-orange body, printed with “S-217 30 mg” on body of capsule in black
5. Warnings and Precautions
5.1 Impaired Ability to Drive or Engage in Other Potentially Hazardous Activities
ZURZUVAE causes driving impairment due to central nervous system (CNS) depressant effects [see Warnings and Precautions (5.2)]. In two driving simulation studies, the driving ability of healthy adults was impaired in a dose-dependent manner following repeat nighttime administration of 30 mg of ZURZUVAE (0.6 times the recommended dose) for five days as well as 50 mg of ZURZUVAE (recommended dose) for seven days [see Clinical Studies (14.2)].
Advise patients not to drive a motor vehicle or engage in other potentially hazardous activities requiring complete mental alertness, such as operating machinery, until at least 12 hours after ZURZUVAE administration for the duration of the 14-day treatment course. Inform patients that they may not be able to assess their own driving competence or the degree of driving impairment caused by ZURZUVAE.
5.2 Central Nervous System Depressant Effects
ZURZUVAE can cause CNS depressant effects such as somnolence and confusion.
In Study 1, 36% of patients who received 50 mg of ZURZUVAE and 6% of patients who received placebo daily developed somnolence. In Study 2, 19% of patients who received another zuranolone capsule formulation (approximately equivalent to 40 mg of ZURZUVAE) and 11% of patients who received placebo daily developed somnolence [see Clinical Studies (14)]. In each clinical study, some ZURZUVAE-treated patients developed confusional state. One of these cases was severe, and was also associated with somnolence, dizziness, and gait disturbance. A higher percentage of ZURZUVAE-treated patients, compared to placebo-treated patients, experienced somnolence, dizziness, or confusion that required dosage reduction, interruption, or discontinuation [see Adverse Reactions (6.1)].
Because ZURZUVAE can cause CNS depressant effects, patients may be at higher risk of falls.
Other CNS depressants such as alcohol, benzodiazepines, opioids, tricyclic antidepressants, or drugs that increase zuranolone concentration, may increase impairment of psychomotor performance or CNS depressant effects such as somnolence, cognitive impairment, and the risk of respiratory depression in ZURZUVAE-treated patients [see Drug Interactions (7)].
To reduce the risk of CNS depressant effects and/or mitigate CNS depressant effects that occurs with ZURZUVAE treatment:
- If patients develop CNS depressant effects, consider dosage reduction or discontinuation of ZURZUVAE [see Dosage and Administration (2.1)].
- If use with another CNS depressant is unavoidable, consider dosage reduction.
- Reduce the ZURZUVAE dosage in patients taking strong CYP3A4 inhibitors [see Dosage and Administration (2.2)].
5.3 Suicidal Thoughts and Behavior
In pooled analyses of placebo-controlled trials of chronically administered antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients and 4,500 pediatric patients, the incidence of suicidal thoughts and behaviors in antidepressant-treated patients age 24 years and younger was greater than in placebo-treated patients. There was considerable variation in risk of suicidal thoughts and behaviors among drugs, but there was an increased risk identified in young patients for most drugs studied. There were differences in absolute risk of suicidal thoughts and behaviors across the different indications, with the highest incidence in patients with major depressive disorder (MDD). The drug-placebo differences in the number of cases of suicidal thoughts and behaviors per 1000 patients treated are provided in Table 1.
| Age Range (years) | Drug-Placebo Difference in Number of Patients with Suicidal Thoughts or Behaviors per 1000 Patients Treated |
|---|---|
|
*ZURZUVAE is not approved in pediatric patients. |
|
| Increases Compared to Placebo | |
| <18 | 14 additional patients |
| 18-24 | 5 additional patients |
| Decreases Compared to Placebo | |
| 25-64 | 1 fewer patient |
ZURZUVAE does not directly affect monoaminergic systems. Consider changing the therapeutic regimen, including discontinuing ZURZUVAE, in patients whose depression becomes worse or who experience emergent suicidal thoughts and behaviors.
5.4 Embryo-fetal Toxicity
Based on findings from animal studies, ZURZUVAE may cause fetal harm when administered to a pregnant woman. In rat studies following exposure during gestation or throughout gestation and lactation, adverse effects on development (fetal malformations, embryofetal and offspring mortality, growth deficits) were observed. In addition, neuronal death was observed in rats exposed to zuranolone during a period of brain development that in humans begins during the third trimester of pregnancy and continues during the first few years after birth [see Use in Specific Populations (8.1, 8.2)].
Advise a pregnant woman of the potential risk to an infant exposed to ZURZUVAE in utero. Advise females of reproductive potential to use effective contraception during treatment with ZURZUVAE and for one week after the final dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Impaired Ability to Drive or Engage in Other Potentially Hazardous Activities [see Warnings and Precautions (5.1)]
- Central Nervous System Depressant Effects [see Warnings and Precautions (5.2)]
- Suicidal Thoughts and Behavior [see Warnings and Precautions (5.3)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ZURZUVAE for the treatment of postpartum depression (PPD) was evaluated in two placebo-controlled clinical studies in 347 women with PPD treated with 50 mg of ZURZUVAE (Study 1), or with another zuranolone capsule formulation approximately equivalent to 40 mg of ZURZUVAE (Study 2) once daily for 14 days [Clinical Studies (14.1)]. The studies included adult patients age 18 to 44 years diagnosed with PPD.
Across PPD clinical studies at all doses studied (Studies 1 and 2), serious adverse reactions included confusional state (1%) [Clinical Studies (14.1)].
In Study 1, the incidence of adverse reactions that led to discontinuation in patients treated with 50 mg of ZURZUVAE and placebo was 2% and 1%, respectively. The most common adverse reaction leading to treatment discontinuation in ZURZUVAE-treated patients was somnolence.
Dosage reduction due to an adverse reaction occurred in 14% of ZURZUVAE-treated patients. The most common adverse reactions leading to dosage reduction in ZURZUVAE-treated patients were somnolence (10%) and dizziness (6%).
The most common adverse reactions (≥5% and greater than placebo) in ZURZUVAE-treated patients were somnolence, dizziness, diarrhea, fatigue, and urinary tract infection.
Table 2 summarizes the adverse reactions that occurred in ≥2% of patients with PPD treated with 50 mg of ZURZUVAE and at a higher incidence than in patients who received placebo in Study 1.
| Adverse Reaction | Placebo
(N=98) (%) | 50 mg of ZURZUVAE
(N=98) (%) |
|---|---|---|
|
1 Somnolence includes sedation and hypersomnia |
||
|
2 Dizziness includes vertigo |
||
|
3 Fatigue includes asthenia |
||
|
4 Abdominal pain includes upper abdominal pain |
||
| Somnolence1 | 6 | 36 |
| Dizziness2 | 9 | 13 |
| Diarrhea | 2 | 6 |
| Fatigue3 | 2 | 5 |
| Urinary tract infection | 4 | 5 |
| Memory impairment | 0 | 3 |
| Abdominal pain4 | 0 | 3 |
| Tremor | 0 | 2 |
| Hypoesthesia | 0 | 2 |
| Muscle twitching | 0 | 2 |
| Myalgia | 0 | 2 |
| COVID-19 | 0 | 2 |
| Anxiety | 1 | 2 |
| Rash | 1 | 2 |
In Study 2, the incidence of adverse reactions that led to discontinuation in patients who received another zuranolone capsule formulation (approximately equivalent to 40 mg of ZURZUVAE) and placebo was 1% and 0%, respectively. The adverse reaction that led to treatment discontinuation was somnolence.
Dosage reduction due to an adverse reaction occurred in 4% of zuranolone-treated patients. The adverse reactions that led to dosage reduction were somnolence and confusional state.
The most common (≥5% and greater than placebo) adverse reactions in zuranolone-treated patients were somnolence, nasopharyngitis, dizziness, fatigue, and diarrhea.
Table 3 summarizes the adverse reactions that occurred in ≥2% of zuranolone-treated patients with PPD and at a higher incidence than in placebo-treated patients in Study 2.
| Adverse Reaction | Placebo
(N=73) (%) | Another Zuranolone Capsule Formulation*
(N=78) (%) |
|---|---|---|
|
1 Somnolence includes sedation |
||
|
2 Nasopharyngitis includes upper respiratory tract infection |
||
|
3 Fatigue includes lethargy |
||
|
* This capsule formulation of zuranolone is approximately equivalent to 40 mg of ZURZUVAE. |
||
| Somnolence1 | 11 | 19 |
| Nasopharyngitis2 | 3 | 9 |
| Dizziness | 6 | 8 |
| Fatigue3 | 1 | 5 |
| Diarrhea | 3 | 5 |
| Dry mouth | 0 | 4 |
| Sinus congestion | 0 | 3 |
| Toothache | 0 | 3 |
7. Drug Interactions
Table 4 displays clinically important drug interactions with ZURZUVAE.
| CNS Depressant Drugs and Alcohol | |
| Clinical Impact | Due to additive pharmacological effects, the concomitant use of CNS depressant drugs, including alcohol, may increase impairment of psychomotor performance or CNS depressant effects. |
| Management | If use with another CNS depressant is unavoidable, consider dosage reduction. Caution should be used when ZURZUVAE is administered in combination with other CNS drugs or alcohol [see Warnings and Precautions (5.2)]. |
| Strong CYP3A4 Inhibitors | |
| Clinical Impact | Concomitant use of ZURZUVAE with a strong CYP3A4 inhibitor increases the exposure of zuranolone [see Clinical Pharmacology (12.3)], which may increase the risk of ZURZUVAE-associated adverse reactions. |
| Management | Reduce the ZURZUVAE dosage when used with a strong CYP3A4 inhibitor [see Dosage and Administration (2.3)]. |
| CYP3A4 Inducers | |
| Clinical Impact | Concomitant use of ZURZUVAE with a CYP3A4 inducer decreases the exposure of zuranolone [see Clinical Pharmacology (12.3)], which may reduce the efficacy of ZURZUVAE. |
| Management | Avoid concomitant use of ZURZUVAE with CYP3A4 inducers [see Dosage and Administration (2.3)]. |
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antidepressants, including ZURZUVAE, during pregnancy. Healthcare providers are encouraged to register patients by calling the National Pregnancy Registry for Antidepressants at 1-844-405-6185 or visiting online at https://womensmentalhealth.org/research/pregnancyregistry/antidepressants/
Risk Summary
Based on findings from animal studies, ZURZUVAE may cause fetal harm. Advise pregnant women of the potential risk to a fetus. Available data on ZURZUVAE use in pregnant women from the clinical development program are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes.
In animals, oral administration of zuranolone to pregnant rats during organogenesis resulted in developmental toxicity, including embryofetal death and fetal malformations, with a no adverse effect level (NOAEL) associated with maternal plasma exposures 7 times greater than in humans at the maximum recommended human dose (MRHD) of 50 mg. Oral administration of zuranolone to rats during pregnancy and lactation resulted in developmental toxicity in the offspring, including, perinatal mortality, at maternal exposures similar to that in humans at the MRHD. Developmental toxicity was observed at doses that were also maternally toxic. Neuronal death was observed in rats exposed to zuranolone during a period of brain development that begins during the third trimester of pregnancy in humans and continues up to a few years after birth.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Oral administration of zuranolone (0, 2.5, 7.5, or 22.5 mg/kg/day) to pregnant rats during organogenesis resulted in increased incidences of fetal malformations, reductions in embryofetal survival, and reduced fetal body weights as well as maternal mortality and sedation at the highest dose. The no effect dose (7.5 mg/kg/day) for adverse effects on embryofetal development was associated with maternal exposures (AUC) approximately 7 times that in humans at the MRHD of 50 mg.
Potential adverse effects of zuranolone on embryofetal development in pregnant rabbits were not adequately assessed.
Oral administration of zuranolone (0, 1, 4, or 10 mg/kg/day) to rats throughout pregnancy and into lactation resulted in increased perinatal mortality and persistent bodyweight reductions in the offspring at the mid and high doses, which also produced maternal mortality and adverse clinical signs. The no-effect dose (1 mg/kg/day) for adverse effects on pre- and postnatal development in rats was associated with maternal exposures (AUC) approximately 2 times that in the humans at the MRHD.
Oral administration of a single dose of zuranolone (0, 2.5, or 7.5 mg/kg) to rats on postnatal day 7 resulted in increased apoptotic neurodegeneration in the brain at the highest dose tested. The no-effect dose (2.5 mg/kg) was associated with plasma exposures (AUC) comparable to that in humans at the MRHD. Brain development on PND 7 in rats corresponds to a period of brain development that begins during the third trimester of pregnancy in humans and continue up to a few years after birth.
8.2 Lactation
Risk Summary
Available data from a clinical lactation study in 14 women indicate that zuranolone is present in low levels in human milk (see Data). There are no data on the effects of zuranolone on a breastfed infant and limited data on the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ZURZUVAE and any potential adverse effects on the breastfed child from ZURZUVAE or from the underlying maternal condition.
Data
A steady-state milk study was conducted in 14 healthy lactating women treated with daily oral administration of 30 mg of ZURZUVAE for 5 days. At steady state (Day 5), the calculated maximum relative infant dose for ZURZUVAE was < 1%. The daily infant dose was low (approximately 0.0013 mg/kg/day), reflecting a mean relative infant dose of 0.357% compared to the maternal dose. Concentrations of ZURZUVAE in breastmilk were below the level of quantification limit (BQL) by 4-6 days after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal studies, ZURZUVAE may cause embryo-fetal harm when administered to a pregnant woman [see Warnings and Precautions (5.4) and Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of ZURZUVAE in pediatric patients have not been established.
8.5 Geriatric Use
PPD is a condition associated with pregnancy; there is no geriatric experience with ZURZUVAE in patients with PPD.
8.6 Hepatic Impairment
Exposure to zuranolone was increased in patients with severe hepatic impairment. The recommended ZURZUVAE dosage in patients with severe hepatic impairment (Child-Pugh C) is lower than patients with normal hepatic function [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
The recommended ZURZUVAE dosage in patients with mild or moderate hepatic impairment (Child-Pugh A or Child-Pugh B) is the same as those with normal hepatic function [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Exposure to zuranolone was increased in patients with moderate (eGFR 30 to 59 mL/min/1.73 m2) and severe (eGFR 15 to 29 mL/min/1.73 m2) renal impairment [see Clinical Pharmacology (12.3)].
The recommended ZURZUVAE dosage in patients with moderate and severe renal impairment is lower than those with normal renal function [see Dosage and Administration (2.4)]. The recommended dosage in patients with mild renal impairment (eGFR 60 to 89 mL/min/1.73 m2) is the same as those in patients with normal renal function. ZURZUVAE has not been studied in patients with eGFR of < 15 mL/min/1.73 m2 or patients requiring dialysis.
9. Drug Abuse and Dependence
9.2 Abuse
Zuranolone has abuse potential with associated risks of misuse, abuse, and substance use disorder including addiction. Abuse is the intentional non-therapeutic use of a drug, even once, for its desired psychological or physiological effects. Misuse is the intentional use, for therapeutic purposes, of a drug by an individual in a way other than prescribed by a health care provider or for whom it was not prescribed. Drug addiction is a cluster of behavioral, cognitive, and physiological phenomena that may include a strong desire to take the drug, difficulties in controlling drug use (e.g., continuing drug use despite harmful consequences, giving a higher priority to drug use than other activities and obligations), and possible tolerance or physical dependence. Individuals with a history of drug abuse or substance use disorders may be at a greater risk of these outcomes with ZURZUVAE.
In a human abuse potential study, single oral doses of 30 mg, 60 mg, and 90 mg of ZURZUVAE (0.6 times, 1.2 times, and 1.8 times the recommended daily dose, respectively) were compared to single oral doses of alprazolam (1.5 mg and 3 mg) and placebo in healthy, nondependent individuals with a history of recreational CNS depressant use. The study demonstrated that ZURZUVAE has dose-dependent abuse potential comparable to alprazolam and greater abuse potential than placebo on positive subjective measures of “drug liking,” “overall drug liking,” “take drug again,” “high,” and “good drug effects.” In the human abuse potential study, dose-dependent, abuse-related adverse reactions, including euphoric mood, feeling drunk, and somnolence, were reported with ZURZUVAE use.
9.3 Dependence
ZURZUVAE may produce physical dependence. Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms after abrupt discontinuation or a significant dose reduction of a drug.
Adverse reactions reported upon discontinuation of zuranolone in healthy subjects who received 50 mg of zuranolone for 5 to 7 days (on the 7th day subjects received 50 mg or 100 mg ) included: insomnia, palpitations, decreased appetite, nightmare, nausea, hyperhidrosis, and paranoia. These adverse reactions indicate a potential for physical dependence with zuranolone. These adverse reactions were mild-to-moderate in severity.
The risk for developing physical dependence and a subsequent withdrawal syndrome upon abrupt ZURZUVAE discontinuation for individuals who take a higher than recommended dosage and/or use ZURZUVAE for a longer duration than recommended [see Dosage and Administration (2.1)] has not been evaluated in clinical studies. However, convulsions were observed in a dog upon abrupt zuranolone discontinuation after dogs were administered zuranolone for 14 days at doses that produced exposures higher than the maximum recommended human dose [see Nonclinical Toxicology (13.2)].
10. Overdosage
There was a case of intentional overdose with ZURZUVAE reported during premarketing clinical trials. The patient took 330 mg (6.5 times the maximum recommended dose) of ZURZUVAE and was reported to be in an altered state of consciousness. The event resolved the next day, following treatment with intravenous fluids.
Overdosage with ZURZUVAE may result in excessive CNS depressant effects such as somnolence and disturbance in consciousness. There is no specific antidote for ZURZUVAE overdosage.
Consider contacting the Poison Help Line (1-800-222-1222) or a medical toxicologist for additional overdosage management recommendations.
11. Zurzuvae Description
ZURZUVAE contains zuranolone, a neuroactive steroid gamma-aminobutyric acid (GABA) A receptor positive modulator. The chemical name is 1-[(3α, 5β)-3-hydroxy-3-methyl-20- oxo-19-norpregnan-21-yl]-1H-pyrazole-4-carbonitrile and it has the following chemical structure:
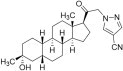
The molecular formula of zuranolone is C25H35N3O2 and the relative molecular mass is 409.57.
Zuranolone is a white to off-white, non-hygroscopic, crystalline solid. It is sparingly soluble in ethyl acetate, methanol, and ethanol; slightly soluble in methyl tert-butyl ether and isopropanol; soluble in tetrahydrofuran and acetone; and practically insoluble in water and n-heptane.
ZURZUVAE is available in 20 mg, 25 mg, and 30 mg capsules for oral administration. Each capsule contains zuranolone as the active ingredient and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, mannitol, microcrystalline cellulose, and sodium stearyl fumarate. The capsule shells contain gelatin, red iron oxide, titanium dioxide, and yellow iron oxide, and are imprinted with black ink (containing ammonium hydroxide, black iron oxide, propylene glycol, and shellac glaze).
12. Zurzuvae - Clinical Pharmacology
12.1 Mechanism of Action
The mechanism of action of zuranolone in the treatment of PPD is not fully understood, but is thought to be related to its positive allosteric modulation of GABAA receptors.
12.2 Pharmacodynamics
12.3 Pharmacokinetics
Zuranolone exposure (Cmax and AUC) increased approximately dose proportionally with doses ranging from 30 mg to 60 mg (1.2 times of the recommended dosage of ZURZUVAE) with a moderate-fat meal (700 calories; 30% fat). Once-daily administration of ZURZUVAE resulted in accumulation of approximately 1.5-fold in systemic exposures and steady state was achieved in 3 to 5 days.
Absorption
Following oral administration, peak zuranolone concentrations occur at 5 to 6 hours (Tmax).
The absolute bioavailability of ZURZUVAE was not evaluated.
Effect of Food
Following administration of 30 mg of ZURZUVAE to healthy subjects, the Cmax increased by approximately 3.5-fold and the AUClast increased by approximately 1.8-fold with a low-fat meal (400 to 500 calories, 25% fat) compared to fasted conditions. The Cmax increased by approximately 4.3-fold and the AUClast increased by approximately 2-fold with a high-fat meal (800 to 1,000 calories, 50% fat) compared to fasted conditions. The Tmax was not impacted by food.
Distribution
The volume of distribution of zuranolone following oral administration is greater than 500 L. The mean blood-to-plasma concentration ratio ranged from 0.54 to 0.58. Plasma protein binding is greater than 99.5%.
Elimination
The terminal half-life of zuranolone is approximately 19.7 to 24.6 hours in an adult population. The mean apparent clearance (CL/F) of zuranolone is 33 L/h.
Metabolism
Zuranolone undergoes extensive metabolism, with CYP3A4 identified as a primary enzyme involved. There were no circulating human metabolites greater than 10% of total drug-related materials and none are considered to contribute to the therapeutic effects of zuranolone.
Excretion
Following oral administration of radiolabeled zuranolone, 45% of the dose was recovered in urine as metabolites with negligible unchanged zuranolone and 41% in feces as metabolites with less than 2% as unchanged zuranolone.
Specific Populations
Racial Groups
Black or African American participants had a 14% higher CL/F compared to participants of other races (Asian, White, or other).
Male and Female Patients, Patients with Renal Impairment, Patients with Hepatic Impairment
Exposures of zuranolone in specific populations are summarized in Figure 1 [see Dosage and Administration (2.4, 2.5) and Use in Specific Populations (8.6, 8.7)].
Figure 1 Effect of Specific Populations on the Pharmacokinetics of Zuranolone

Data shown for participants ≥65 years relative to younger participants (18 to 45 years); female participants relative to male participants; renal and hepatic impairment relative to participants with normal renal and hepatic function, respectively. Hepatic impairment: Mild (Child-Pugh Class A); moderate (Child-Pugh Class B); severe (Child-Pugh Class C). Renal impairment: Mild (eGFR 60-89 mL/min/1.73m2); moderate (eGFR 30-59 mL/min/1.73m2); severe (eGFR <30 mL/min/1.73m2 and not on dialysis). CI = confidence interval; GMR = geometric mean ratio
Drug Interaction Studies
The effect of co-administered drugs on the pharmacokinetics of zuranolone is summarized in Figure 2 [see Dosage and Administration (2.3) and Drug Interactions (7)].
Figure 2 Effect of Co-Administered Drugs on the Pharmacokinetics of Zuranolone

Data shown are zuranolone plus co-administered drug relative to zuranolone alone. CI = confidence interval; GMR = geometric mean ratio; * clinically significant drug interaction
ZURZUVAE did not affect the pharmacokinetics of alprazolam or ethanol. Increased impairment of psychomotor performance was observed when ZURZUVAE was co-administered with alprazolam or ethanol [see Warnings and Precautions (5.2) and Drug Interactions (7)].
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Oral administration of zuranolone in a 26-week carcinogenicity study in transgenic mice (0, 10, 30, or 100 mg/kg/day), and in a 104-week carcinogenicty study in rats (0, 2, 6, or 20 mg/kg/day in males and 0, 0.2, 0.6, or 1.5 mg/kg/day in females) was not associated with increases in tumors in either species. Plasma exposures (AUC) in rats at the highest dose tested were approximately 4 times that in humans at the maximum recommended human dose (MRHD) of 50 mg.
Mutagenesis
Zuranolone was not genotoxic when tested in an in vitro microbial mutagenicity (Ames) assay, an in vitro chromosome aberration assay in Chinese hamster ovary cells, and an in vivo bone marrow micronucleus assay in rats.
Impairment of Fertility
Oral administration of zuranolone (0, 3, 10, or 30 mg/kg/day) to male rats prior to and during mating with untreated females resulted in increased post-implantation loss and a corresponding decrease in the number of viable embryos at the high dose, which was also paternally toxic. There were no adverse effects on fertility or sperm parameters. Adverse effects on reproduction were not observed when males were remated after a 6-week treatment-free period. The no effect dose (10 mg/kg/day) for male reproductive toxicity was associated with a plasma zuranolone exposure (AUC) of approximately 4 times the human exposure at the MRHD. Oral administration of zuranolone (0, 1, 3, or 10 mg/kg/day) to female rats prior to and throughout mating and continuing through early gestation resulted in disruption of estrous cyclicity at the high dose, but there were no adverse effects on fertility or early embryonic development. The no-effect dose (3 mg/kg/day) for female reproductive toxicity was associated with exposures approximately 4 times that in humans at the MRHD.
13.2 Animal Toxicology and/or Pharmacology
Death was observed in 1 dog four days after repeat dosing with 2.5 mg/kg for 9 months was stopped. Death/euthanasia in 2 dogs was also observed 2 to 4 days after dosing with 2.5 mg/kg for 14 days was stopped. Convulsions were observed in 1 dog three days after repeat dosing with 2.5 mg/kg for 14 days was stopped. Plasma exposure (AUC) at the no-effect dose in dogs was approximately 3 times the human exposure at the MRHD.
14. Clinical Studies
14.1 Postpartum Depression
The efficacy of ZURZUVAE for the treatment of postpartum depression (PPD) in adults was demonstrated in two randomized, placebo-controlled, double-blind, multicenter studies (Study 1, NCT04442503 and Study 2, NCT02978326) in women with PPD who met the Diagnostic and Statistical Manual of Mental Disorders criteria for a major depressive episode (DSM-5) with onset of symptoms in the third trimester or within 4 weeks of delivery. In these studies, concomitant use of existing oral antidepressants was allowed for patients taking a stable dose of oral antidepressant for at least 30 days before baseline. These studies included patients with HAMD-17 scores ≥ 26 at baseline.
In Study 1, patients received 50 mg of ZURZUVAE (N=98) or placebo (N=97) once daily in the evening with fat-containing food for 14 days, with the option to reduce the dosage based on tolerability to 40 mg once daily of ZURZUVAE or placebo. The patients were followed for a minimum of 4 weeks after the 14-day treatment course.
In Study 2, patients received another zuranolone capsule formulation (approximately equivalent to 40 mg of ZURZUVAE) (N=76) or placebo (N=74) once daily in the evening with food for 14 days. The patients were followed for a minimum of 4 weeks after the 14-day treatment course.
Baseline Demographics and Disease Characteristics
In Studies 1 and 2, the baseline demographic and disease characteristics of patients were similar between the ZURZUVAE and placebo groups. In Study 1, patients had a mean age of 30 years (range 19 to 44 years); were 70% White, 22% Black or African American, 1% Asian, and 7% were other races; and 38% were of Hispanic or Latino ethnicity. Baseline use of stable oral antidepressants was reported in 15% of patients. In Study 2, patients had a mean age of 28 years (range 18 to 44 years); were 56% White, 41% Black or African American, 1% Asian, and 2% were other races; and 23% were of Hispanic or Latino ethnicity. Baseline use of stable oral antidepressants was reported in 19% of patients.
The primary endpoint for Studies 1 and 2 was the change from baseline in depressive symptoms as measured by the HAMD-17 total score at Day 15. In these studies, patients in the ZURZUVAE groups experienced statistically significantly greater improvement on the primary endpoint compared to patients in the placebo groups, as shown in Table 5. For Study 1, the key secondary endpoints included change from baseline in HAMD-17 total score at Days 3, 28, and 45.
|
HAMD-17: 17-item Hamilton depression rating scale; SD: standard deviation; LS: least squares; SE: standard error; CI: confidence interval |
|||||
|
*This capsule formulation of zuranolone is approximately equivalent to 40 mg of ZURZUVAE. |
|||||
| Study Number | Treatment Group | N | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo- subtracted Difference
(95% CI) |
| 1 | 50 mg of ZURZUVAE | 98 | 28.6 (2.49) | -15.6 (0.82) | -4.0 (-6.3, -1.7) |
| Placebo | 97 | 28.8 (2.34) | -11.6 (0.82) | ||
| 2 | Zuranolone (another capsule formulation)* | 76 | 28.4 (2.09) | -17.8 (1.04) | -4.2 (-6.9, -1.5) |
| Placebo | 74 | 28.8 (2.32) | -13.6 (1.07) | ||
Subgroup analyses of the primary endpoint did not suggest differences in response to 50 mg of ZURZUVAE for age, race, or BMI.
The time course of response for 50 mg ZURZUVAE compared to placebo for Study 1 is shown in Figure 3.
Figure 3 Mean Change from Baseline in HAM-D Total Score Over Time (Days) in Women with PPD in Study 1
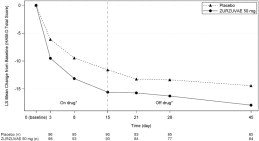
*Both phases were double-blind.
14.2 Effects on Driving
Two randomized, double-blind, placebo- and active-controlled, four-way crossover studies (Study 3 and Study 4) evaluated the effects of nighttime ZURZUVAE administration on next-morning driving performance, 9 hours after dosing, using a computer-based driving simulation.
Study 3
In Study 3, 50 mg of ZURZUVAE was administered for six consecutive nights and on the seventh night a single dose of 50 mg or 100 mg (two times the recommended dose) was administered. The primary driving performance outcome measure was the change in Standard Deviation of Lateral Position (SDLP) (a measure of driving impairment) in the ZURZUVAE group compared to the placebo group on Days 2 and 8 (after a single dose and repeat doses, respectively).
Study 3 included 67 healthy participants. The median age was 45 years old (age ranged from 22 to 81 years old; 7 participants were ≥ 65 years of age); there were 38 males and 29 females; 88% were White, 5% were Black or African American, 3% were Asian, and 5% were other races; and 12% were of Hispanic/Latino ethnicity.
A single 50 mg dose of ZURZUVAE caused statistically significant impairment in next-morning driving performance compared to placebo. Statistically significant effects on driving were also observed on Day 8 following daily administration of 50 mg of ZURZUVAE. Administration of 100 mg of ZURZUVAE (twice the maximum recommended dose) on the final night increased impairment in driving ability [see Warnings and Precautions (5.1)].
The exposure-response analysis for driving impairment in Study 3 suggested that the projected mean placebo-adjusted SDLP at 12 hours post-dose would be less than the threshold associated with driving impairment.
Study 4
In Study 4, 30 mg of ZURZUVAE (0.6 times the maximum recommended daily dose) was administered for four consecutive nights and on the fifth night a single dose of 30 mg or 60 mg (1.2 times the recommended daily dose) was administered. The primary driving performance outcome measure was the change in SDLP in the ZURZUVAE group compared to the placebo group on Days 2 and 6 (after a single dose and repeat doses, respectively).
Study 4 included 60 participants; 60% and 40% were male and female, respectively; the median age was 41 years old (range was 22 to 62 years old); 90% were White, 5% were Black or African American, 3% were Asian, and 2% were other races; and 15% were of Hispanic/Latino ethnicity.
A single 30 mg dose of ZURZUVAE caused a statistically significant impairment in next-morning driving performance compared to placebo. The mean effect on driving performance was not statistically significantly different following 30 mg of ZURZUVAE compared to placebo on Day 6; however, driving ability was impaired in some participants taking ZURZUVAE. Administration of 60 mg of ZURZUVAE (1.2 times the maximum recommended dose) on the final night caused statistically significant impairment in next-morning driving performance compared to placebo [see Warnings and Precautions (5.1)].
16. How is Zurzuvae supplied
How Supplied
ZURZUVAE (zuranolone) is supplied as 20 mg, 25 mg, and 30 mg capsules as follows:
| Capsule Strength | Capsule Colors | Capsule Markings | Packaging Configuration | NDC |
|---|---|---|---|---|
| 20 mg | light-orange cap ivory to light-yellow body | Black “S-217 20 mg” on body | Bottle of 14 | 64406-029-01 |
| 25 mg | light-orange cap light-orange body | Black “S-217 25 mg” on body | Bottle of 14 | 64406-030-01 |
| Blister pack of 28 | 64406-030-02 | |||
| 30 mg | orange cap light-orange body | Black “S-217 30 mg” on body | Bottle of 14 | 64406-031-01 |
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Impaired Ability to Drive or Engage in Other Potentially Hazardous Activities
Inform patients that ZURZUVAE causes driving impairment. Advise patients to take ZURZUVAE in the evening. Advise patients not to drive a motor vehicle or engage in other potentially hazardous activities requiring complete mental alertness, such as operating machinery, until at least 12 hours after ZURZUVAE administration. Warn patients that they may not be able to assess their own ability to drive or the degree of impairment caused by ZURZUVAE [see Warnings and Precautions (5.1)].
Central Nervous System Depressant Effects
Inform patients that ZURZUVAE can cause somnolence, dizziness, sedation, and confusion. Advise patients to use caution if taking ZURZUVAE in combination with alcohol, other CNS depressants, or medications that increase the concentration of ZURZUVAE [see Warnings and Precautions (5.2), Adverse Reactions (6.1) and Drug Interactions (7)].
Suicidal Thoughts and Behavior
Advise patients to look for the emergence of suicidal thoughts and behaviors and instruct them to report such symptoms to the healthcare provider immediately [see Warnings and Precautions (5.3)].
Administration Instructions
Inform patients to take ZURZUVAE with fat-containing food [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)].
Instruct patients that if a daily dose is missed, the missed dose should be taken the next day as scheduled. Patients should be advised not to take extra capsules to make up for the missed dose [see Dosage and Administration (2.1, 2.5)].
Abuse, Misuse, and Physical Dependence
Advise patients that ZURZUVAE has abuse potential with associated risks of misuse, abuse, and substance use disorder including addiction and that ZURZUVAE is associated with the potential for physical dependence [see Drug Abuse and Dependence (9.2, 9.3)].
Pregnancy
Advise pregnant women and females of reproductive potential of the potential risk to a fetus and to notify their healthcare provider if they become pregnant or intend to become pregnant during treatment with ZURZUVAE. Advise female patients of reproductive potential to use effective contraception during treatment with ZURZUVAE and for one week after the final dose [see Warnings and Precautions (5.4), Use in Specific Populations (8.1, 8.3)].
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ZURZUVAE during pregnancy [see Use in Specific Populations (8.1)].
Manufactured for:
Biogen Inc.
225 Binney Street
Cambridge, MA 02142 USA
ZURZUVAE is a trademark of Sage Therapeutics, Inc.
© 2024 Sage Therapeutics and Biogen
|
This Medication Guide has been approved by the U.S. Food and Drug Administration. |
Revised: 07/2024 |
||
| MEDICATION GUIDE
ZURZUVAE™ (zur-ZOO-vay) (zuranolone) capsules, for oral use, CIV |
|||
|
What is the most important information I should know about ZURZUVAE?
|
|||
| What is ZURZUVAE?
ZURZUVAE is a prescription medicine used to treat adults with postpartum depression (PPD). It is not known if ZURZUVAE is safe and effective for use in children. ZURZUVAE is a federal controlled substance (C-IV) because it contains zuranolone that can be abused or lead to dependence. Keep ZURZUVAE in a safe place to protect it from theft. Do not sell or give away ZURZUVAE because it may harm others and is against the law. |
|||
Before taking ZURZUVAE, tell your healthcare provider about all of your medical conditions, including if you:
|
|||
| Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. ZURZUVAE and some medicines may interact with each other and cause serious side effects. ZURZUVAE may affect the way other medicines work and other medicines may affect the way ZURZUVAE works. Especially tell your healthcare provider if you take:
|
|||
| Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. Your healthcare provider will decide if other medicines can be taken with ZURZUVAE. | |||
How should I take ZURZUVAE?
|
|||
What should I avoid while taking ZURZUVAE?
|
|||
| See “What is the most important information I should know about ZURZUVAE?” | |||
| What are the possible side effects of ZURZUVAE?
ZURZUVAE may cause serious side effects, including:
|
|||
|
|
||
| The most common side effects of ZURZUVAE include: | |||
|
|
||
| These are not all of the possible side effects of ZURZUVAE. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
How should I store ZURZUVAE?
|
|||
| Keep ZURZUVAE and all medicines out of the reach of children. | |||
| General information about the safe and effective use of ZURZUVAE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ZURZUVAE for a condition for which it was not prescribed. Do not give ZURZUVAE to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ZURZUVAE that is written for health professionals. |
|||
| What are the ingredients in ZURZUVAE?
Active ingredient: zuranolone Inactive ingredients: ZURZUVAE capsules contain colloidal silicon dioxide, croscarmellose sodium, mannitol, microcrystalline cellulose, and sodium stearyl fumarate. The capsule shells contain gelatin, red iron oxide, titanium dioxide, and yellow iron oxide. The imprinting ink contains ammonium hydroxide, black iron oxide, propylene glycol, and shellac glaze. Manufactured for: Biogen Inc., 225 Binney Street, Cambridge, MA 02142 ZURZUVAE is a trademark of Sage Therapeutics, Inc. For more information about ZURZUVAE go to www.ZURZUVAE.com or call 1-844-987-9882. |
|||
PRINCIPAL DISPLAY PANEL - 30 mg Bottle Carton
NDC 64406-031-01
Rx Only
ZURZUVAE™
CIV
(zuranolone) capsules
30 mg
For oral use
Attention: Dispense
with accompanying
Medication Guide.
14 Capsules

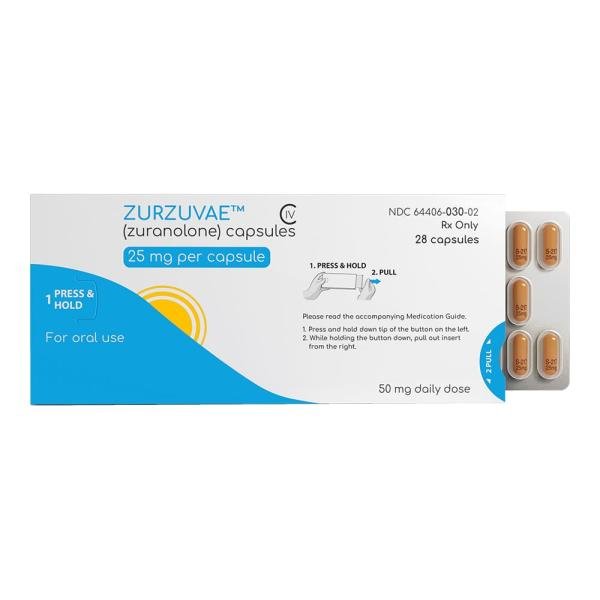
PRINCIPAL DISPLAY PANEL - 25 mg Blister Pack Carton
ZURZUVAE™
CIV
(zuranolone) capsules
25 mg per capsule
For oral use
Attention: Dispense
with accompanying
Medication Guide.
28 Capsules
NDC 64406-030-02
Rx Only
Contains a 14-day blister
pack with 28 capsules.
Each capsule contains
25 mg of zuranolone.
50 mg daily dose
21709-4


| ZURZUVAE
zuranolone capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ZURZUVAE
zuranolone capsule |
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||
| ZURZUVAE
zuranolone capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Biogen MA Inc. (121376230) |
Frequently asked questions
More about Zurzuvae (zuranolone)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: miscellaneous antidepressants
- Breastfeeding
- En español