Sunosi: Package Insert / Prescribing Info
Package insert / product label
Generic name: solriamfetol
Dosage form: tablet, film coated
Drug class: Miscellaneous central nervous system agents
Medically reviewed by Drugs.com. Last updated on Jul 12, 2023.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Drug Abuse and Dependence
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
SUNOSI® (solriamfetol) tablets, for oral use, CIV
Initial U.S. Approval: 2019
Indications and Usage for Sunosi
SUNOSI is a dopamine and norepinephrine reuptake inhibitor (DNRI) indicated to improve wakefulness in adult patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnea (OSA). (1)
Limitations of Use
SUNOSI is not indicated to treat the underlying airway obstruction in OSA. Ensure that the underlying airway obstruction is treated (e.g., with continuous positive airway pressure (CPAP)) for at least one month prior to initiating SUNOSI for excessive daytime sleepiness. Modalities to treat the underlying airway obstruction should be continued during treatment with SUNOSI. SUNOSI is not a substitute for these modalities. (1)
Sunosi Dosage and Administration
- Administer once daily upon awakening. Avoid administration within 9 hours of planned bedtime because of the potential to interfere with sleep. (2.2)
- Starting dose for patients with narcolepsy: 75 mg once daily. (2.3)
- Starting dose for patients with OSA: 37.5 mg once daily. (2.4)
- Dose may be increased at intervals of at least 3 days. (2.3, 2.4)
- Maximum dose is 150 mg once daily. (2.3, 2.4)
- Renal impairment (2.5, 8.6, 12.3):
- Moderate impairment: Starting dose is 37.5 mg once daily.
- May increase to 75 mg once daily after at least 7 days.
- Severe impairment: Starting dose and maximum dose is 37.5 mg once daily.
- End stage renal disease (ESRD): Not recommended.
- Moderate impairment: Starting dose is 37.5 mg once daily.
Dosage Forms and Strengths
Tablets: 75 mg (functionally scored) and 150 mg. (3)
Contraindications
- Concurrent treatment with a monoamine oxidase inhibitor (MAOI) or use of an MAOI within the preceding 14 days. (4)
Warnings and Precautions
- Blood Pressure and Heart Rate Increases: Measure heart rate and blood pressure prior to initiating and periodically throughout treatment. Control hypertension before and during therapy. Avoid use in patients with unstable cardiovascular disease, serious heart arrhythmias, or other serious heart problems. (5.1)
- Psychiatric Symptoms: Use caution in treating patients with a history of psychosis or bipolar disorders. Consider dose reduction or discontinuation of SUNOSI if psychiatric symptoms develop. (5.2)
Adverse Reactions/Side Effects
Most common adverse reactions (≥ 5% and greater than placebo): headache, nausea, decreased appetite, insomnia, and anxiety. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Axsome Therapeutics, Inc. at 1-800-484-1672 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2023
Full Prescribing Information
1. Indications and Usage for Sunosi
SUNOSI is indicated to improve wakefulness in adult patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnea (OSA) [see Clinical Studies (14)].
Limitations of Use
SUNOSI is not indicated to treat the underlying airway obstruction in OSA. Ensure that the underlying airway obstruction is treated (e.g., with continuous positive airway pressure (CPAP)) for at least one month prior to initiating SUNOSI for excessive daytime sleepiness. Modalities to treat the underlying airway obstruction should be continued during treatment with SUNOSI. SUNOSI is not a substitute for these modalities.
2. Sunosi Dosage and Administration
2.1 Important Considerations Prior to Initiating Treatment
Prior to initiating treatment with SUNOSI, ensure blood pressure is adequately controlled [see Warnings and Precautions (5.1)].
2.2 General Administration Instructions
Administer SUNOSI orally upon awakening with or without food. Avoid taking SUNOSI within 9 hours of planned bedtime because of the potential to interfere with sleep if taken too late in the day.
SUNOSI 75 mg tablets are functionally scored tablets that can be split in half (37.5 mg) at the score line.
2.3 Dosage in Narcolepsy
Initiate SUNOSI at 75 mg once daily in adults with narcolepsy. The recommended dose range for SUNOSI is 75 mg to 150 mg once daily. Based on efficacy and tolerability, the dosage of SUNOSI may be doubled at intervals of at least 3 days. The maximum recommended dose is 150 mg once daily. Dosages above 150 mg daily do not confer increased effectiveness sufficient to outweigh dose-related adverse reactions [see Warnings and Precautions (5.1)].
2.4 Dosage in OSA
Initiate SUNOSI at 37.5 mg once daily in adults with OSA. The recommended dosage range for SUNOSI is 37.5 mg to 150 mg once daily. Based on efficacy and tolerability, the dosage of SUNOSI may be doubled at intervals of at least 3 days. The maximum recommended dosage is 150 mg once daily. Dosages above 150 mg daily do not confer increased effectiveness sufficient to outweigh dose-related adverse reactions [see Warnings and Precautions (5.1)].
2.5 Dosage Recommendations in Patients with Renal Impairment
Moderate renal impairment (eGFR 30-59 mL/min/1.73 m2): Initiate dosing at 37.5 mg once daily. Based on efficacy and tolerability, dose may be increased to a maximum of 75 mg once daily after at least 7 days [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
Severe renal impairment (eGFR 15-29 mL/min/1.73 m2): Administer 37.5 mg once daily. The maximum recommended daily dose is 37.5 mg [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
End Stage Renal Disease (eGFR <15 mL/min/1.73 m2): SUNOSI is not recommended for use in patients with ESRD [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
SUNOSI 75 mg – (75 mg solriamfetol equivalent to 89.3 mg of the hydrochloride salt) dark yellow oblong tablet with "75" debossed on one side and a functional score line on the opposite side.
SUNOSI 150 mg – (150 mg solriamfetol equivalent to 178.5 mg of the hydrochloride salt) yellow oblong tablet with "150" debossed on one side.
4. Contraindications
SUNOSI is contraindicated in patients receiving concomitant treatment with monoamine oxidase (MAO) inhibitors, or within 14 days following discontinuation of monoamine oxidase inhibitor, because of the risk of hypertensive reaction [see Drug Interactions (7.1)].
5. Warnings and Precautions
5.1 Blood Pressure and Heart Rate Increases
SUNOSI increases systolic blood pressure, diastolic blood pressure, and heart rate in a dose-dependent fashion [see Adverse Reactions (6.1)].
Epidemiological data show that chronic elevations in blood pressure increase the risk of major adverse cardiovascular events (MACE), including stroke, heart attack, and cardiovascular death. The magnitude of the increase in absolute risk is dependent on the increase in blood pressure and the underlying risk of MACE in the population being treated. Many patients with narcolepsy and OSA have multiple risk factors for MACE, including hypertension, diabetes, hyperlipidemia, and high body mass index (BMI).
Assess blood pressure and control hypertension before initiating treatment with SUNOSI. Monitor blood pressure regularly during treatment and treat new-onset hypertension and exacerbations of pre-existing hypertension. Exercise caution when treating patients at higher risk of MACE, particularly patients with known cardiovascular and cerebrovascular disease, pre-existing hypertension, and patients with advanced age. Use caution with other drugs that increase blood pressure and heart rate [see Drug Interactions (7.2)].
Periodically reassess the need for continued treatment with SUNOSI. If a patient experiences increases in blood pressure or heart rate that cannot be managed with dose reduction of SUNOSI or other appropriate medical intervention, consider discontinuation of SUNOSI.
Patients with moderate or severe renal impairment may be at a higher risk of increases in blood pressure and heart rate because of the prolonged half-life of SUNOSI [see Dosage and Administration (2.5), Clinical Pharmacology (12.3)].
5.2 Psychiatric Symptoms
Psychiatric adverse reactions have been observed in clinical trials with SUNOSI, including anxiety, insomnia, and irritability [see Adverse Reactions (6.1)].
SUNOSI has not been evaluated in patients with psychosis or bipolar disorders. Exercise caution when treating patients with SUNOSI who have a history of psychosis or bipolar disorders.
Patients with moderate or severe renal impairment may be at a higher risk of psychiatric symptoms because of the prolonged half-life of SUNOSI [see Dosage and Administration (2.5), Clinical Pharmacology (12.3)].
Patients treated with SUNOSI should be observed for the possible emergence or exacerbation of psychiatric symptoms. If psychiatric symptoms develop in association with the administration of SUNOSI, consider dose reduction or discontinuation of SUNOSI.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the label:
- Blood Pressure and Heart Rate Increases [see Warnings and Precautions (5.1)]
- Psychiatric Symptoms [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of SUNOSI has been evaluated in 930 patients (ages 18 to 75 years) with narcolepsy or OSA. Among these patients, 396 were treated with SUNOSI in the 12-week placebo-controlled trials at doses of 37.5 mg (OSA only), 75 mg, and 150 mg once daily. Information provided below is based on the pooled 12-week placebo-controlled studies in patients with narcolepsy or OSA.
Most Common Adverse Reactions
The most common adverse reactions (incidence ≥ 5% and greater than placebo) reported more frequently with the use of SUNOSI than placebo in either the narcolepsy or OSA populations were headache, nausea, decreased appetite, anxiety, and insomnia.
Table 1 presents the adverse reactions that occurred at a rate of ≥ 2% and more frequently in SUNOSI-treated patients than in placebo-treated patients in the narcolepsy population.
| * “Insomnia” includes insomnia, initial insomnia, middle insomnia, and terminal insomnia. “Anxiety” includes anxiety, nervousness, and panic attack. “Headache” includes headache, tension headache, and head discomfort. “Nausea” includes nausea and vomiting. | ||
| Narcolepsy | ||
| System Organ Class | Placebo
N = 108 (%) | SUNOSI
N = 161 (%) |
| Metabolism and Nutrition Disorders
Decreased appetite |
1 |
9 |
| Psychiatric Disorders
Insomnia* Anxiety* |
4 1 |
5 6 |
| Nervous System Disorders Headache* |
7 |
16 |
| Cardiac Disorders
Palpitations |
1 |
2 |
| Gastrointestinal Disorders
Nausea* Dry mouth Constipation |
4 2 1 |
7 4 3 |
Table 2 presents the adverse reactions that occurred at a rate of ≥ 2% and more frequently in SUNOSI-treated patients than in placebo-treated patients in the OSA population.
| * “Anxiety” includes anxiety, nervousness, and panic attack. “Nausea” includes nausea and vomiting. “Abdominal pain” includes abdominal pain, abdominal pain upper, and abdominal discomfort. | ||
| OSA | ||
| System Organ Class | Placebo
N = 118 (%) | SUNOSI
N = 235 (%) |
| Metabolism and Nutrition Disorders
Decreased appetite |
1 |
6 |
| Psychiatric Disorders
Anxiety* Irritability |
1 0 |
4 3 |
| Nervous System Disorders Dizziness |
1 |
2 |
| Cardiac Disorders
Palpitations |
0 |
3 |
| Gastrointestinal Disorders
Nausea* Diarrhea Abdominal pain* Dry mouth |
6 1 2 2 |
8 4 3 3 |
| General Disorders and Administration Site Conditions
Feeling jittery Chest discomfort |
0 0 |
3 2 |
| Skin and Subcutaneous Tissue Disorders
Hyperhidrosis |
0 |
2 |
Other Adverse Reactions Observed During the Premarketing Evaluation of SUNOSI
Other adverse reactions of < 2% incidence but greater than placebo are shown below. The following list does not include adverse reactions: 1) already listed in previous tables or elsewhere in the labeling, 2) for which a drug cause was remote, 3) which were so general as to be uninformative, or 4) which were not considered to have clinically significant implications.
Narcolepsy population:
Psychiatric disorders: agitation, bruxism, irritability
Respiratory, thoracic and mediastinal disorders: cough
Skin and subcutaneous tissue disorders: hyperhidrosis
General disorders and administration site conditions: feeling jittery, thirst, chest discomfort, chest pain
Investigations: weight decreased
OSA population:
Psychiatric disorders: bruxism, restlessness
Nervous system disorders: disturbance in attention, tremor
Respiratory, thoracic and mediastinal disorders: cough, dyspnea
Gastrointestinal disorders: constipation, vomiting
Investigations: weight decreased
Dose-Dependent Adverse Reactions
In the 12-week placebo-controlled clinical trials that compared doses of 37.5 mg, 75 mg, and 150 mg daily of SUNOSI to placebo, the following adverse reactions were dose-related: headache, nausea, decreased appetite, anxiety, diarrhea, and dry mouth (Table 3).
| * In OSA only. ** “Headache” includes headache, tension headache, and head discomfort. “Nausea” includes nausea and vomiting. |
||||
| Placebo
N = 226 (%) | SUNOSI 37.5 mg
N = 58* (%) | SUNOSI 75 mg
N = 120 (%) | SUNOSI 150 mg
N = 218 (%) |
|
| Headache** | 8 | 7 | 9 | 13 |
| Nausea** | 5 | 7 | 5 | 9 |
| Decreased appetite | 1 | 2 | 7 | 8 |
| Anxiety | 1 | 2 | 3 | 7 |
| Dry mouth | 2 | 2 | 3 | 4 |
| Diarrhea | 2 | 2 | 4 | 5 |
Adverse Reactions Resulting in Discontinuation of Treatment
In the 12-week placebo-controlled clinical trials, 11 of the 396 patients (3%) who received SUNOSI discontinued because of an adverse reaction compared to 1 of the 226 patients (< 1%) who received placebo. The adverse reactions resulting in discontinuation that occurred in more than one SUNOSI-treated patient and at a higher rate than placebo were: anxiety (2/396; < 1%), palpitations (2/396; < 1%), and restlessness (2/396; < 1%).
Increases in Blood Pressure and Heart Rate
SUNOSI’s effects on blood pressure and heart rate are summarized below. Table 4 shows maximum mean changes in blood pressure and heart rate recorded at sessions where the Maintenance of Wakefulness Test (MWT) was administered [see Clinical Studies (14)]. Table 5 summarizes 24-hour ambulatory blood pressure monitoring (ABPM) and ambulatory heart rate monitoring performed in the outpatient setting.
| SBP = systolic blood pressure; DBP = diastolic blood pressure; HR = heart rate * For study weeks 1, 4, and 12, SBP, DBP, and HR were assessed pre-dose and every 1-2 hours for 10 hours after test drug administration. For all time points at all visits, the mean change from baseline was calculated, by indication and dose, for all patients with a valid assessment. The table shows, by indication and dose, the mean changes from baseline for the week and time point with the maximal change in SBP, DBP, and HR. ** The maximum recommended daily dose is 150 mg. Dosages above 150 mg daily do not confer increased effectiveness sufficient to outweigh dose-related adverse reactions. |
||||||
| Placebo | SUNOSI 37.5 mg | SUNOSI 75 mg | SUNOSI 150 mg | SUNOSI 300 mg** |
||
| Narcolepsy
STUDY 1 | n | 52 | - | 51 | 49 | 53 |
| SBP | 3.5 (0.7, 6.4) | 3.1 (0.1, 6.0) | 4.9 (1.7, 8.2) | 6.8 (3.2, 10.3) | ||
| n | 23 | - | 47 | 49 | 53 | |
| DBP | 1.8 (-1.8, 5.5) | 2.2 (0.2, 4.1) | 4.2 (2.0, 6.5) | 4.2 (1.5, 6.9) | ||
| n | 48 | - | 26 | 49 | 53 | |
| HR | 2.3 (-0.1, 4.7) | 3.7 (0.4, 6.9) | 4.9 (2.3, 7.6) | 6.5 (3.9, 9.0) | ||
| OSA
STUDY 2 | n | 35 | 17 | 54 | 103 | 35 |
| SBP | 1.7 (-1.4, 4.9) | 4.6 (-1.1, 10.2) | 3.8 (1.2, 6.4) | 2.4 (0.4, 4.4) | 4.5 (1.1, 7.9) | |
| n | 99 | 17 | 17 | 107 | 91 | |
| DBP | 1.4 (-0.1, 2.9) | 1.9 (-2.3, 6.0) | 3.2 (-0.9, 7.3) | 1.8 (0.4, 3.2) | 3.3 (1.8, 4.8) | |
| n | 106 | 17 | 51 | 102 | 91 | |
| HR | 1.7 (0.1, 3.3) | 1.9 (-1.9, 5.7) | 3.3 (0.6, 6.0) | 2.9 (1.4, 4.4) | 4.5 (3.0, 6.0) | |
|
SBP = systolic blood pressure; DBP = diastolic blood pressure; HR = heart rate * Number of patients who had at least 50% valid ABPM readings. |
||||||
| Placebo | SUNOSI 37.5 mg | SUNOSI 75 mg | SUNOSI 150 mg | SUNOSI 300 mg** |
||
| Narcolepsy
STUDY 1 | n* | 46 | 44 | 44 | 40 | |
| SBP | -0.4 (-3.1, 2.4) | - | 1.6 (-0.4, 3.5) | -0.5 (-2.1, 1.1) | 2.4 (0.5, 4.3) | |
| DBP | -0.2 (-1.9, 1.6) | - | 1.0 (-0.4, 2.5) | 0.8 (-0.4, 2.0) | 3.0 (1.4, 4.5) | |
| HR | 0.0 (-1.9, 2.0) | - | 0.2 (-2.1, 2.4) | 1.0 (-1.2, 3.2) | 4.8 (2.3, 7.2) | |
| OSA
STUDY 2 | n* | 92 | 43 | 49 | 96 | 84 |
| SBP | -0.2 (-1.8, 1.4) | 1.8 (-1.1, 4.6) | 2.6 (0.02, 5.3) | -0.2 (-2.0, 1.6) | 2.8 (-0.1, 5.8) | |
| DBP | 0.2 (-0.9, 1.3) | 1.4 (-0.4, 3.2) | 1.5 (-0.04, 3.1) | -0.1 (-1.1, 1.0) | 2.4 (0.5, 4.4) | |
| HR | -0.4 (-1.7, 0.9) | 0.4 (-1.4, 2.2) | 1.0 (-0.9, 2.81) | 1.7 (0.5, 2.9) | 1.6 (0.3, 2.9) | |
6.2 Postmarketing Experience
The following adverse reactions have been reported during post-approval use of SUNOSI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Immune system disorders: Hypersensitivity (rash erythematous, rash [unspecified], and urticaria).
7. Drug Interactions
7.1 Monoamine Oxidase (MAO) Inhibitors
Do not administer SUNOSI concomitantly with MAOIs or within 14 days after discontinuing MAOI treatment. Concomitant use of MAO inhibitors and noradrenergic drugs may increase the risk of a hypertensive reaction. Potential outcomes include death, stroke, myocardial infarction, aortic dissection, ophthalmological complications, eclampsia, pulmonary edema, and renal failure [see Contraindications (4)].
7.2 Drugs that Increase Blood Pressure and/or Heart Rate
Concomitant use of SUNOSI with other drugs that increase blood pressure and/or heart rate has not been evaluated, and such combinations should be used with caution [see Warnings and Precautions (5.1)].
7.3 Dopaminergic Drugs
Dopaminergic drugs that increase levels of dopamine or that bind directly to dopamine receptors might result in pharmacodynamic interactions with SUNOSI. Interactions with dopaminergic drugs have not been evaluated with SUNOSI. Use caution when concomitantly administering dopaminergic drugs with SUNOSI.
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to SUNOSI during pregnancy. Healthcare providers are encouraged to register pregnant patients, or pregnant women may enroll themselves in the registry by calling 1-877-283-6220 or contacting the company at www.SunosiPregnancyRegistry.com.
Risk Summary
Available data from case reports are not sufficient to determine drug-associated risks of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproductive studies, oral administration of solriamfetol during organogenesis caused maternal and fetal toxicities in rats and rabbits at doses ≥ 4 and 5 times and was teratogenic at doses 19 and ≥ 5 times, respectively, the maximum recommended human dose (MRHD) of 150 mg based on mg/m2 body surface area. Oral administration of solriamfetol to pregnant rats during pregnancy and lactation at doses ≥ 7 times the MRHD based on mg/m2 body surface area resulted in maternal toxicity and adverse effects on fertility, growth, and development in offspring (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2% to 4% and 15% to 20%, respectively.
Animal Data
Solriamfetol was administered orally to pregnant rats during the period of organogenesis at 15, 67, and 295 mg/kg/day, which are approximately 1, 4, and 19 times the MRHD based on mg/m2 body surface area. Solriamfetol at ≥ 4 times the MRHD caused maternal toxicity that included hyperactivity, significant decreases in body weight, weight gain, and food consumption. Fetal toxicity at these maternally toxic doses included increased incidence of early resorption and post-implantation loss, and decreased fetal weight. Solriamfetol was teratogenic at 19 times the MRHD; it increased the incidence of fetal malformations that included severe sternebrae mal-alignment, hindlimb rotation, bent limb bones, and situs inversus. This dose was also maternally toxic. The no-adverse-effect level for malformation is 4 times and for maternal and embryofetal toxicity is approximately 1 times the MRHD based on mg/m2 body surface area.
Solriamfetol was administered orally to pregnant rabbits during the period of organogenesis at 17, 38, and 76 mg/kg/day, which are approximately 2, 5, and 10 times the MRHD based on mg/m2 body surface area. Solriamfetol at 10 times the MRHD caused maternal toxicity of body weight loss and decreased food consumption. Solriamfetol was teratogenic at ≥ 5 times the MRHD, it caused fetal skeletal malformation (slight-to-moderate sternebrae mal-alignment) and decreased fetal weight. The no-adverse-effect level for malformation and fetal toxicity is approximately 2 times and for maternal toxicity is approximately 5 times the MRHD based on mg/m2 body surface area.
Solriamfetol was administered orally to pregnant rats during the period of organogenesis from gestation day 7 through lactation day 20 post-partum, at 35, 110, and 350 mg/kg/day, which are approximately 2, 7, and 22 times the MRHD based on mg/m2 body surface area. At ≥ 7 times the MRHD, solriamfetol caused maternal toxicity that included decreased body weight gain, decreased food consumption, and hyperpnea. At these maternally toxic doses, fetal toxicity included increased incidence of stillbirth, postnatal pup mortality, and decreased pup weight. Developmental toxicity in offspring after lactation day 20 included decreased body weight, decreased weight gain, and delayed sexual maturation. Mating and fertility of offspring were decreased at maternal doses 22 times the MRHD without affecting learning and memory. The no-adverse-effect level for maternal and developmental toxicity is approximately 2 times the MRHD based on mg/m2 body surface area.
8.2 Lactation
Risk Summary
Available data from a lactation study in 6 women indicate that solriamfetol is present in human milk. The daily infant dose is 0.112 mg/kg (based on nominal infant weight of 6 kg) and the relative infant dose (RID) is approximately 5.5% of the maternal weight-adjusted dosage (see Data). Data are insufficient to determine effects of solriamfetol on a breastfed infant or its effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SUNOSI and any potential adverse effects on the breastfed infant from SUNOSI or from the underlying maternal condition.
Clinical Considerations
Monitor infants exposed to SUNOSI for signs of agitation, insomnia, and reduced weight gain.
Data
A single-dose milk and plasma lactation study was conducted in 6 healthy adult lactating women who were between 10 days and 52 weeks postpartum and were administered a single oral 150 mg dose of SUNOSI. The cumulative median amount excreted in breast milk was 0.67 mg over 72 hours, which is about 5.5% of the maternal dose on a weight-adjusted basis. Of the total amount of solriamfetol excreted in breast milk over 72 hours, approximately 78% and 98% were excreted by 8 and 24 hours, respectively, with an apparent mean elimination half-life in breast milk of about 5 hours.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established. Clinical studies of SUNOSI in pediatric patients have not been conducted.
8.5 Geriatric Use
Of the total number of patients in the narcolepsy and OSA clinical studies treated with SUNOSI, 13% (123/930) were 65 years of age or over.
No clinically meaningful differences in safety or effectiveness were observed between elderly and younger patients.
Solriamfetol is predominantly eliminated by the kidney. Because elderly patients are more likely to have decreased renal function, dosing may need to be adjusted based on eGFR in these patients. Consideration should be given to the use of lower doses and close monitoring in this population [see Dosage and Administration (2.5)].
8.6 Renal Impairment
Dosage adjustment is not required for patients with mild renal impairment (eGFR 60-89 mL/min/1.73 m2). Dosage adjustment is recommended for patients with moderate to severe renal impairment (eGFR 15-59 mL/min/1.73 m2). SUNOSI is not recommended for patients with end stage renal disease (eGFR <15 mL/min/1.73 m2) [see Dosage and Administration (2.5), Warnings and Precautions (5.1, 5.2), Clinical Pharmacology (12.3)].
9. Drug Abuse and Dependence
9.2 Abuse
SUNOSI has potential for abuse. Abuse is the intentional non-therapeutic use of a drug, even once, to achieve a desired psychological or physiological effect. The abuse potential of SUNOSI 300 mg, 600 mg, and 1200 mg (two, four, and eight times the maximum recommended dose, respectively) was assessed relative to phentermine, 45 mg and 90 mg, (a Schedule IV controlled substance) in a human abuse potential study in individuals experienced with the recreational use of stimulants. Results from this clinical study demonstrated that SUNOSI produced Drug Liking scores similar to or lower than phentermine. In this crossover study, elevated mood was reported by 2.4% of placebo-treated subjects, 8 to 24% of SUNOSI-treated subjects, and 10 to 18% of phentermine-treated subjects. A ‘feeling of relaxation’ was reported in 5% of placebo-treated subjects, 5 to 19% of SUNOSI-treated subjects and 15 to 20% of phentermine-treated subjects.
Physicians should carefully evaluate patients for a recent history of drug abuse, especially those with a history of stimulant (e.g., methylphenidate, amphetamine, or cocaine) or alcohol abuse, and follow such patients closely, observing them for signs of misuse or abuse of SUNOSI (e.g., incrementation of doses, drug-seeking behavior).
9.3 Dependence
In a long-term safety and maintenance of efficacy study, the effects of abrupt discontinuation of SUNOSI were evaluated following at least 6 months of SUNOSI use in patients with narcolepsy or OSA. The effects of abrupt discontinuation of SUNOSI were also evaluated during the two-week safety follow-up periods in the Phase 3 studies. There was no evidence that abrupt discontinuation of SUNOSI resulted in a consistent pattern of adverse events in individual subjects that was suggestive of physical dependence or withdrawal.
10. Overdosage
A specific reversal agent for SUNOSI is not available. Hemodialysis removed approximately 21% of a 75 mg dose in end stage renal disease patients. Overdoses should be managed with primarily supportive care, including cardiovascular monitoring.
Consult with a Certified Poison Control Center at 1-800-222-1222 for latest recommendations.
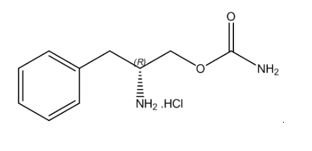
11. Sunosi Description
SUNOSI contains solriamfetol, a dopamine and norepinephrine reuptake inhibitor (DNRI). Solriamfetol is a phenylalanine derivative with the systematic name (R)-2-amino-3-phenylpropylcarbamate hydrochloride.
The molecular formula is C10H15N2O2Cl, and the molecular weight is 230.69.
The chemical structure is:
Solriamfetol hydrochloride is a white to off-white solid that is freely soluble in water.
SUNOSI tablets are intended for oral administration. Each 75 mg SUNOSI film-coated tablet contains 75 mg solriamfetol (equivalent to 89.3 mg solriamfetol hydrochloride). Each 150 mg SUNOSI film-coated tablet contains 150 mg solriamfetol (equivalent to 178.5 mg solriamfetol hydrochloride). The inactive ingredients are hydroxypropyl cellulose and magnesium stearate. In addition, the film coating contains: iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
12. Sunosi - Clinical Pharmacology
12.1 Mechanism of Action
The mechanism of action of solriamfetol to improve wakefulness in patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnea is unclear. However, its efficacy could be mediated through its activity as a dopamine and norepinephrine reuptake inhibitor (DNRI).
12.2 Pharmacodynamics
Solriamfetol binds to the dopamine transporter and norepinephrine transporter with low affinity (Ki=14.2 µM and 3.7 µM, respectively), and inhibits the reuptake of dopamine and norepinephrine with low potency (IC50 =2.9 μM and 4.4 μM, respectively). Solriamfetol has no appreciable binding affinity for the serotonin transporter (Ki=81.5 µM) and does not inhibit serotonin reuptake (IC50> 100 μM). Solriamfetol has no appreciable binding affinity to dopamine, serotonin, norepinephrine, GABA, adenosine, histamine, orexin, benzodiazepine, muscarinic acetylcholine, or nicotinic acetylcholine receptors.
Cardiac Electrophysiology
The effect of solriamfetol 300 mg and 900 mg (twice and six times the maximum recommended dose, respectively) on the QTc interval was evaluated in a randomized, double-blind, placebo-, and positive-controlled (moxifloxacin 400 mg), 4-period, crossover study in 60 healthy subjects. A large increase in heart rate was observed in both solriamfetol treatment groups (mean change from baseline in HR of 21 and 27 bpm in the 300 and 900 mg groups, respectively, compared with 8 bpm in the placebo group). These heart rate effects impact the interpretability of the QTc effects, particularly in the 900 mg group. In this study, solriamfetol 300 mg did not prolong the QTcF interval to a clinically relevant extent.
12.3 Pharmacokinetics
Solriamfetol exhibits linear kinetics over the dose range of 42 to 1008 mg (approximately 0.28 to 6.7 times the maximum recommended dosage). Steady state is reached in 3 days, and once‑daily administration is expected to result in minimal accumulation (1.06 times single‑dose exposure).
Absorption
The oral bioavailability of solriamfetol is approximately 95%. Peak plasma concentration of solriamfetol occurs at a median Tmax of 2 hours (range 1.25 to 3.0 hours) post-dose under fasted conditions.
Effect of Food
Ingestion of solriamfetol with a high-fat meal resulted in minimal change in Cmax and AUCinf; however, a delay of approximately 1 hour in Tmax was observed.
Distribution
The apparent volume of distribution of solriamfetol is approximately 199 L. Plasma protein binding ranged from 13.3% to 19.4% over solriamfetol concentration range of 0.059 to 10.1 mcg/mL in human plasma. The mean blood‑to‑plasma concentration ratio ranged from 1.16 to 1.29.
Elimination
Solriamfetol exhibits first‑order elimination after oral administration. The apparent mean elimination half‑life is about 7.1 hours.
Metabolism
Solriamfetol is minimally metabolized in humans.
Excretion
Approximately 95% of the dose was recovered in urine as unchanged solriamfetol, and 1% or less of the dose was recovered as the minor inactive metabolite N‑acetyl solriamfetol in a mass balance study. Renal clearance (18.2 L/h) represented the majority of apparent total clearance (19.5 L/h). Active tubular secretion is likely involved in the renal elimination of the parent drug.
Specific Populations
Population PK analysis indicated that age, gender, and race do not have clinically relevant effects on the pharmacokinetics of solriamfetol. No dose adjustments were made in clinical studies that enrolled patients ages 65 and above.
Patients with Renal Impairment
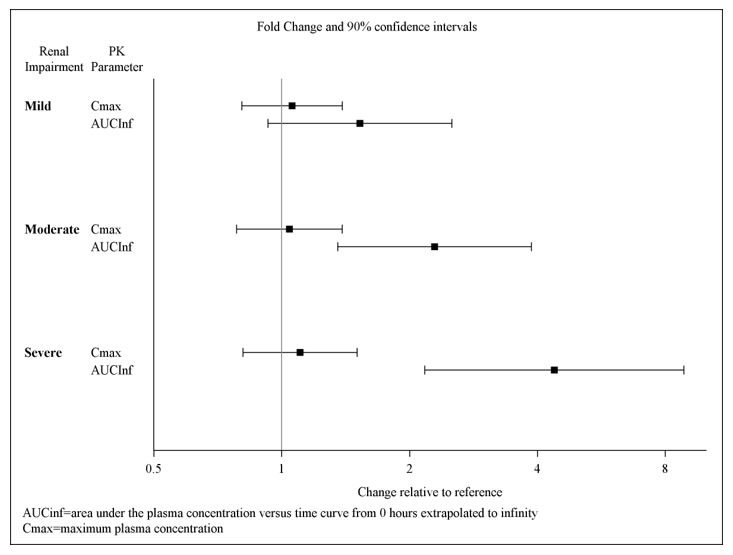
Exposures to solriamfetol in patients with renal impairment compared to subjects with normal renal function (eGFR ≥ 90 mL/min/1.73 m2) are summarized in Figure 1. The half‑life of solriamfetol was increased approximately 1.2‑, 1.9‑, and 3.9‑fold in patients with mild (eGFR 60‑89 mL/min/1.73 m2), moderate (eGFR 30–59 mL/min/1.73 m2), or severe (eGFR <30 mL/min/1.73 m2) renal impairment, respectively. Exposure (AUC) and half-life of solriamfetol was significantly increased in patients with ESRD (eGFR <15 mL/min/1.73 m2) [see Use in Specific Populations (8.6)]. An average of 21% of solriamfetol was removed by hemodialysis. In general, median Tmax values were not affected by renal impairment.
Figure 1: Effect of Renal Impairment on Solriamfetol Pharmacokinetics
Drug Interaction Studies
In Vitro Studies
CYP and UGT Enzymes: Solriamfetol was minimally metabolized in vitro. Solriamfetol is not an inhibitor of CYPs 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, or 3A4. It does not induce CYP1A2, 2B6, 3A4, or UGT1A1 enzymes at clinically relevant concentrations.
Transporter Systems: Solriamfetol is a low-avidity substrate of OCT2, MATE1, OCTN1, and OCTN2. Solriamfetol is a weak inhibitor of OCT2 (IC50 of 146 μM) and MATE1 (IC50 of 211 μM), and is not an inhibitor of OCT1, MATE2-K, OCTN1, or OCTN2. Solriamfetol does not appear to be a substrate or inhibitor of P-gp, BCRP, OATP1B1, OATP1B3, OAT1, or OAT3.
Based on in vitro data, clinically significant PK drug interactions with major CYPs and transporters are not expected in patients taking SUNOSI.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Solriamfetol did not increase the incidence of tumors in rats or mice treated orally for up to 101 and 104 weeks at 35, 80, and 200 mg/kg/day (rat), and 20, 65, and 200 mg/kg/day (mouse), respectively. These doses are approximately 2, 6, and 18 times (rat), and 0.4, 2.6, and 7 times (mouse) the MRHD based on AUC.
Mutagenesis
Solriamfetol was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay or clastogenic in the in vitro mammalian chromosomal aberration assay or in the in vivo mouse bone marrow micronucleus assay.
Impairment of Fertility
Solriamfetol did not affect fertility or sperm parameters when administered orally to male rats for 8 weeks at doses of 35 and 110 mg/kg/day, which are approximately 2 and 7 times the MRHD, based on mg/m2 body surface area. At 350 mg/kg/day, which is approximately 22 times the MRHD based on mg/m2 body surface area, solriamfetol decreased sperm count and sperm concentration without affecting fertility.
Solriamfetol did not affect fertility when administered orally to female rats for 2 weeks premating, during mating, and through gestation day 7 at 15, 67, and 295 mg/kg/day, which are approximately 1, 4, and 19 times the MRHD, based on mg/m2 body surface area.
14. Clinical Studies
14.1 Narcolepsy
The efficacy of SUNOSI in improving wakefulness and reducing excessive daytime sleepiness was demonstrated in a 12-week, multi-center, randomized, double-blind, placebo-controlled, parallel-group study (Study 1; NCT02348593) in adult patients with a diagnosis of narcolepsy according to the ICSD-3 or DSM-5 criteria.
Wakefulness and sleepiness were assessed using the Maintenance of Wakefulness Test (MWT) and the Epworth Sleepiness Scale (ESS). The MWT measures an individual’s ability to remain awake during the daytime in a darkened, quiet environment. Patients were instructed to remain awake for as long as possible during 40-minute test sessions, and sleep latency was determined as the mean number of minutes patients could remain awake in the first four test sessions. The ESS is an 8-item questionnaire by which patients rate their perceived likelihood of falling asleep during usual daily life activities. Change in overall symptom severity was assessed using the Patient Global Impression of Change (PGIc) scale. The PGIc is a 7-point patient-reported scale by which patients rate their symptom change since the beginning of the study. Responses range from “very much improved” to “very much worse.” The co-primary efficacy endpoints were change from baseline in MWT and ESS at Week 12. A pre-specified secondary endpoint was percentage of subjects reported as improved (minimally, much, or very much) at Week 12 by PGIc.
A total of 239 patients with narcolepsy were randomized to receive SUNOSI 75 mg, 150 mg, or 300 mg (two times the maximum recommended daily dose), or placebo once daily. Patients randomized to the 150-mg dose received 75 mg for the first 3 days before increasing to 150 mg.
Demographic and baseline disease characteristics were similar for the SUNOSI and placebo groups. Median age was 34 years (range 18 to 70 years), 65% were female, 80% were Caucasian, 14% were African American, and 3% were Asian. Approximately 51% of patients had cataplexy.
Compared to the placebo group, patients randomized to 150 mg SUNOSI showed statistically significant improvements on the MWT (treatment effect difference: 7.7 minutes, Table 6) and on the ESS (treatment effect difference: 3.8 points, Table 7) at Week 12. These effects were apparent at Week 1 and consistent with the results at Week 12. The change on percentage of subjects reported as improved by PGIc was also statistically significant compared with placebo. There were trends toward improvement in the SUNOSI 75-mg treatment group (Tables 6 and 7); however, these changes were not statistically significant. There was no evidence of differential efficacy in patients with cataplexy and patients without cataplexy. Examination of subgroups by age, race, and sex did not suggest differences in response.
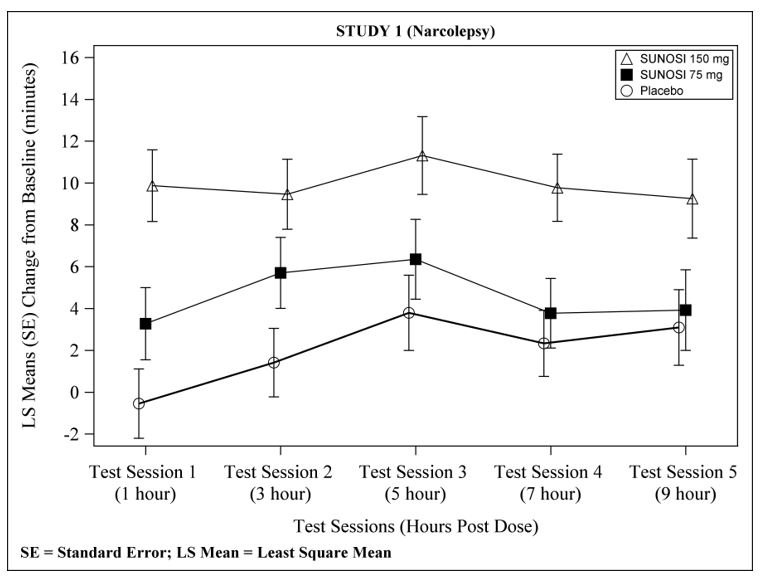
At Week 12, 150 mg of SUNOSI demonstrated improvements in wakefulness compared to placebo as assessed in test sessions 1 (approximately 1 hour post-dose) through 5 (approximately 9 hours post-dose) of the MWT (Figure 2). Nighttime sleep as measured with polysomnography was not affected by the use of SUNOSI in Study 1.
Figure 2: Maintenance of Wakefulness Test Improvements in Test Sessions 1 through 5 in Patients with Narcolepsy in Study 1 at Week 12
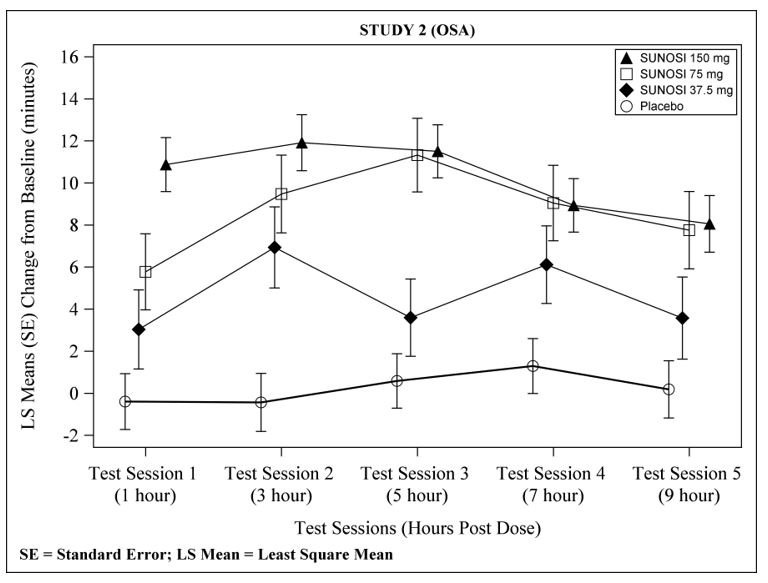
14.2 Obstructive Sleep Apnea (OSA)
The efficacy of SUNOSI in improving wakefulness and reducing excessive daytime sleepiness in patients with OSA was demonstrated in a 12-week multi-center, randomized, double-blind, placebo-controlled study (Study 2; NCT02348606) in adults diagnosed with OSA according to ICSD-3 criteria. The co-primary efficacy endpoints were change from baseline in MWT and ESS at Week 12; a pre-specified secondary endpoint was percentage of subjects reported as improved (minimally, much, or very much) at Week 12 by PGIc.
A total of 476 patients with OSA were randomized to receive SUNOSI 37.5 mg, 75 mg, 150 mg, or 300 mg (two times the maximum recommended daily dose), or placebo once daily. Patients randomized to the 150-mg dose received 75 mg for the first 3 days before increasing to 150 mg.
Demographic and baseline disease characteristics were similar for the SUNOSI and placebo groups. Median age was 55 years (range 20 to 75 years), 37% were female, 76% were Caucasian, 19% were African American, and 4% were Asian.
Compared to the placebo group, patients randomized to 37.5 mg, 75 mg, and 150 mg SUNOSI showed statistically significant improvements on the MWT (treatment effect difference: 4.5 minutes, 8.9 minutes, and 10.7 minutes respectively; Table 6) and ESS (treatment effect difference: 1.9 points, 1.7 points, and 4.5 points respectively; Table 7) at Week 12. These effects were apparent at Week 1 and consistent with the results at Week 12. The change on percentage of subjects reported as improved by PGIc was also statistically significant compared with placebo at the 75 mg and 150 mg doses. Examination of subgroups by age, race, and sex did not suggest differences in response.
At Week 12, 37.5 mg, 75 mg, and 150 mg of SUNOSI all demonstrated improvements in wakefulness compared to placebo as assessed in test sessions 1 (approximately 1 hour post-dose) through 5 (approximately 9 hours post-dose) of the MWT (Figure 3). Nighttime sleep as measured with polysomnography was not affected by the use of SUNOSI in Study 2. Patients’ compliance with a primary OSA therapy device was similar across the placebo and SUNOSI treatment groups at baseline, and did not change during the 12-week study period in any treatment group.
Figure 3: Maintenance of Wakefulness Test Improvements in Test Sessions 1 through 5 in Patients with OSA in Study 2 at Week 12
| SD = standard deviation; SE = standard error; LS Mean = least square mean; CI = confidence interval Maximum possible MWT score is 40 minutes. A positive change represents improvement. Difference from Placebo = LS Mean Difference between change from baseline between active drug and placebo. * Dose that was statistically significantly superior to placebo after adjusting for multiplicity. |
||||||
| Indication/ Study | Treatment Group (N) | Baseline Mean (SD) | LS Mean Change from Baseline at Week 12 (SE) | Difference from Placebo (95% CI) |
||
| Narcolepsy
STUDY 1 | Placebo (58) | 6.2 (5.7) | 2.1 (1.3) | - | ||
| SUNOSI 75 mg (59) | 7.5 (5.4) | 4.7 (1.3) | 2.6 (-1.0, 6.3) | |||
| SUNOSI 150 mg* (55) | 7.9 (5.7) | 9.8 (1.3) | 7.7 (4.0, 11.3) | |||
| OSA
STUDY 2 | Placebo (114) | 12.6 (7.1) | 0.2 (1.0) | - | ||
| SUNOSI 37.5 mg* (56) | 13.6 (8.1) | 4.7 (1.4) | 4.5 (1.2, 7.9) | |||
| SUNOSI 75 mg* (58) | 12.4 (6.9) | 9.1 (1.4) | 8.9 (5.6, 12.4) | |||
| SUNOSI 150 mg* (116) | 12.5 (7.2) | 11.0 (1.0) | 10.7 (8.1, 13.4) | |||
| SD = standard deviation; SE = standard error; LS Mean = least square mean; CI = confidence interval Scores range from 0 to 24 with higher scores indicating more severe sleepiness. A negative change represents improvement. Difference from placebo = LS mean difference between change from baseline between SUNOSI and placebo. * Dose that was statistically significantly superior to placebo after adjusting for multiplicity. |
||||||
| Indication/ Study | Treatment Groups (N) | Baseline Score Mean (SD) | LS Mean Change from Baseline at Week 12 (SE) | Difference from Placebo (95% CI) |
||
| Narcolepsy
STUDY 1 | Placebo (58) | 17.3 (2.9) | -1.6 (0.7) | - | ||
| SUNOSI 75 mg (59) | 17.3 (3.5) | -3.8 (0.7) | -2.2 (-4.0, -0.3) | |||
| SUNOSI 150 mg* (55) | 17.0 (3.6) | -5.4 (0.7) | -3.8 (-5.6, -2.0) | |||
| OSA
STUDY 2 | Placebo (114) | 15.6 (3.3) | -3.3 (0.5) | - | ||
| SUNOSI 37.5 mg* (56) | 15.1 (3.5) | -5.1 (0.6) | -1.9 (-3.4, -0.3) | |||
| SUNOSI 75 mg* (58) | 15.0 (3.5) | -5.0 (0.6) | -1.7 (-3.2, -0.2) | |||
| SUNOSI 150 mg* (116) | 15.1 (3.4) | -7.7 (0.4) | -4.5 (-5.7, -3.2) | |||
14.3 Maintenance of Efficacy in Narcolepsy and OSA
The maintenance of effect of SUNOSI in improving wakefulness and reducing excessive daytime sleepiness in patients with narcolepsy and OSA was assessed in two randomized-withdrawal, placebo-controlled studies, Study 3 (NCT02348619) and Study 4 (NCT02348632).) and Study 4 (NCT02348632).
Study 3 was a 6-week, multi-center, double-blind, placebo-controlled, randomized-withdrawal study in 174 adult patients with a diagnosis of OSA. The co-primary efficacy endpoints were change from the beginning to the end of the randomized withdrawal period in MWT and ESS. During a 2-week, open-label titration phase, patients were started on SUNOSI 75 mg once daily, and were titrated to the maximum tolerable dose between 75 mg and 300 mg per day (two times the maximum recommended daily dose). Patients were continued on this dose for a 2-week stable-dose phase. At the end of the stable-dose phase, 124 patients who reported “much” or “very much” improvement on the PGIc and who showed improvements on the MWT and ESS entered a double-blind withdrawal phase and were randomized 1:1 to either continue SUNOSI at the dose received in the stable-dose phase or switch to placebo. Compared to patients who remained on SUNOSI, patients randomized to placebo experienced statistically significant worsening of sleepiness as measured by the MWT and ESS (Table 8).
Study 4 was a 52-week, open-label study in 638 patients with either narcolepsy or OSA who had completed a prior trial. During a 2-week, open-label titration phase, patients were started on SUNOSI 75 mg once daily, and were titrated to the maximum tolerable dose between 75 mg and 300 mg per day (two times the maximum recommended daily dose). Patients remained on this dose during a subsequent open-label treatment period of either 38 (for patients previously enrolled in Study 1 or Study 2) or 50 (all others) weeks. A 2-week randomized-withdrawal period was incorporated into the study. After 6 months of stable-dose treatment, 282 patients (79 with narcolepsy; 203 with OSA) entered the randomized-withdrawal period. Patients were randomized 1:1 to either continue to receive SUNOSI at the dose received in the maintenance phase or to switch to placebo. The primary efficacy endpoint was change from the beginning to the end of the randomized-withdrawal period in ESS. Compared to patients who remained on SUNOSI, patients randomized to placebo experienced statistically significant worsening of sleepiness as measured by the ESS (Table 8).
| SD = standard deviation; SE = standard error; LS Mean = least square mean; CI = confidence interval For MWT, maximum possible score is 40 minutes; negative changes indicate worsening. For ESS, scores range from 0 to 24; positive changes indicate worsening. * Statistically significantly superior to placebo after adjusting for multiplicity. |
||||||
| Indication/ Study | Endpoint | Treatment Groups (N) | Beginning of Randomized Withdrawal Period (Baseline) Mean (SD) | LS Mean Change from Baseline (SE) | Difference from Placebo (95% CI) |
|
| OSA
STUDY 3 | MWT (minutes) | Placebo (62) | 29.0 (9.9) | -12.1 (1.3) | ||
| SUNOSI* (60) | 31.7 (9.2) | -1.0 (1.4) | 11.2 (7.8, 14.6) | |||
| ESS Score | Placebo (62) | 5.9 (3.8) | 4.5 (0.7) | |||
| SUNOSI* (60) | 6.4 (4.4) | -0.1 (0.7) | -4.6 (-6.4, -2.8) | |||
| OSA and Narcolepsy
STUDY 4 | ESS Score | Placebo (141) | 7.8 (5.0) | 5.3 (0.4) | ||
| SUNOSI* (139) | 7.3 (5.3) | 1.6 (0.4) | -3.7 (-4.8, -2.7) | |||
16. How is Sunosi supplied
How Supplied
SUNOSI is packaged in 30-count white, high density polyethylene (HDPE) bottles.
SUNOSI tablets, 75 mg - dark yellow oblong tablet with "75" debossed on one side and a functional score line on the opposite side.
NDC 81968-350-01: Bottles of 30 with child-resistant closure
SUNOSI tablets, 150 mg - yellow oblong tablet with "150" debossed on one side.
NDC 81968-351-01: Bottles of 30 with child-resistant closure
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Potential for Abuse and Dependence
Advise patients that SUNOSI is a federally controlled substance because it has the potential to be abused [see Drug Abuse and Dependence (9)]. Advise patients to keep their medication in a secure place and to dispose of unused SUNOSI as recommended in the Medication Guide.
Primary OSA Therapy Use
Inform patients that SUNOSI is not indicated to treat the airway obstruction in OSA and they should use a primary OSA therapy, such as CPAP, as prescribed to treat the underlying obstruction [see Indications and Usage (1)]. SUNOSI is not a substitute for primary OSA therapy.
Blood Pressure and Heart Rate Increases
Instruct patients that SUNOSI can cause elevations of their blood pressure and pulse rate and that they should be monitored for such effects [see Warnings and Precautions (5.1)].
Psychiatric Symptoms
Instruct patients to contact their healthcare provider if they experience, anxiety, insomnia, irritability, agitation, or signs of psychosis or bipolar disorders [see Warnings and Precautions (5.2)].
Lactation
Advise breastfeeding patients using SUNOSI to monitor infants for signs of agitation, insomnia, and reduced weight [see Use in Specific Populations (8.2)].
For more information, visit www.SUNOSI.com
Distributed by:
Axsome Therapeutics, Inc.
New York, NY 10007
For patent information: www.axsome.com/IP
© 2023 Axsome Therapeutics, Inc.
SUN-USPI-004.000-20230630
| This Medication Guide has been approved by the U.S. Food and Drug Administration. SUN-USMG-003.000-20230630 | Revised: 06/2023 |
|
MEDICATION GUIDE
SUNOSI® (suh-NOH-see) |
|
|
What is SUNOSI? SUNOSI is a prescription medicine used to improve wakefulness in adults with excessive daytime sleepiness that is associated with narcolepsy or obstructive sleep apnea (OSA).
SUNOSI is a federally controlled substance (CIV) because it contains solriamfetol that can be a target for people who abuse prescription medicines or street drugs. Keep SUNOSI in a safe place to protect it from theft. Never give your SUNOSI to anyone else, because it may cause death or harm them. Selling or giving away SUNOSI may harm others and is against the law. Tell your healthcare provider if you have ever abused or been dependent on alcohol, prescription medicines or street drugs. |
|
| Do not take SUNOSI if you are taking or have stopped taking within the past 14 days a medicine used to treat depression called a monoamine oxidase inhibitor (MAOI). | |
Before taking SUNOSI, tell your healthcare provider about all your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription or over-the-counter medicines, vitamins, or herbal supplements. SUNOSI and some other medicines may affect each other causing possible serious side effects. SUNOSI may affect the way other medicines work and other medicines may affect the way SUNOSI works. Especially tell your healthcare provider if you take a medicine used to treat depression called a monoamine oxidase inhibitor (MAOI). Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|
How should I take SUNOSI?
|
|
| What are the possible side effects of SUNOSI? SUNOSI may cause serious side effects, including:
The most common side effects of SUNOSI include:
These are not all the possible side effects of SUNOSI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store SUNOSI?
Keep SUNOSI and all medicines out of the reach of children. |
|
| General information about the safe and effective use of SUNOSI.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use SUNOSI for a condition for which it was not prescribed. Do not give SUNOSI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about SUNOSI that is written for health professionals. |
|
| What are the ingredients in SUNOSI?
Active Ingredient: solriamfetol Inactive Ingredients: hydroxypropyl cellulose and magnesium stearate. In addition, the film coating contains: iron oxide yellow, polyethylene glycol, polyvinyl alcohol, titanium dioxide, and talc. Distributed by: Axsome Therapeutics, Inc. New York, NY 10007 |
|




| SUNOSI
solriamfetol tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| SUNOSI
solriamfetol tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Axsome Therapeutics, Inc. (033333109) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Siegfried Malta Ltd | 647169494 | MANUFACTURE(81968-350, 81968-351) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AndersonBrecon Inc. | 053217022 | LABEL(81968-350, 81968-351) , PACK(81968-350, 81968-351) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AndersonBrecon (UK) | 762771269 | LABEL(81968-350, 81968-351) , PACK(81968-350, 81968-351) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Siegfried AG,Zofingen,Switzerland | 482824026 | API MANUFACTURE(81968-350, 81968-351) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Almac Sciences (Ireland) Limited | 985822621 | ANALYSIS(81968-350, 81968-351) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| UFAG Laboratorien AG | 486383151 | ANALYSIS(81968-350, 81968-351) | |
Frequently asked questions
More about Sunosi (solriamfetol)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (80)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- FDA approval history
- Drug class: miscellaneous central nervous system agents
- Breastfeeding
- En español