Mitoxantrone Injection Concentrate: Package Insert / Prescribing Info
Package insert / product label
Generic name: mitoxantrone hydrochloride
Dosage form: injection, solution, concentrate
Drug class: Antibiotics / antineoplastics
J Code (medical billing code): J9293 (Per 5 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jan 23, 2025.
On This Page
- Description
- Clinical Pharmacology
- Clinical Studies
- Indications and Usage
- Contraindications
- Warnings
- Precautions
- Patient Counseling Information
- Drug Interactions
- Adverse Reactions/Side Effects
- Overdosage
- Dosage and Administration
- References
- How Supplied/Storage and Handling
- Storage and Handling
- Medication Guide

WARNING
Mitoxantrone Injection, USP (concentrate) should be administered under the supervision of a physician experienced in the use of cytotoxic chemotherapy agents.
Mitoxantrone Injection, USP (concentrate) should be given slowly into a freely flowing intravenous infusion. It must never be given subcutaneously, intramuscularly, or intra-arterially. Severe local tissue damage may occur if there is extravasation during administration. (See ADVERSE REACTIONS, General, Cutaneous and DOSAGE AND ADMINISTRATION, Preparation and Administration Precautions).
NOT FOR INTRATHECAL USE. Severe injury with permanent sequelae can result from intrathecal administration. (See WARNINGS, General)
Except for the treatment of acute nonlymphocytic leukemia, mitoxantrone therapy generally should not be given to patients with baseline neutrophil counts of less than 1,500 cells/mm3. In order to monitor the occurrence of bone marrow suppression, primarily neutropenia, which may be severe and result in infection, it is recommended that frequent peripheral blood cell counts be performed on all patients receiving mitoxantrone.
Cardiotoxicity
Congestive heart failure (CHF), potentially fatal, may occur either during therapy with mitoxantrone or months to years after termination of therapy. Cardiotoxicity risk increases with cumulative mitoxantrone dose and may occur whether or not cardiac risk factors are present. Presence or history of cardiovascular disease, radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, or use of other cardiotoxic drugs may increase this risk. In cancer patients, the risk of symptomatic CHF was estimated to be 2.6% for patients receiving up to a cumulative dose of 140 mg/m2. To mitigate the cardiotoxicity risk with mitoxantrone, prescribers should consider the following:
All Patients
- •
- All patients should be assessed for cardiac signs and symptoms by history, physical examination, and ECG prior to start of mitoxantrone therapy.
- •
- All patients should have baseline quantitative evaluation of left ventricular ejection fraction (LVEF) using appropriate methodology (ex. Echocardiogram, multi-gated radionuclide angiography (MUGA), MRI, etc.).
Multiple Sclerosis Patients
- •
- MS patients with a baseline LVEF below the lower limit of normal should not be treated with mitoxantrone.
- •
- MS patients should be assessed for cardiac signs and symptoms by history, physical examination and ECG prior to each dose.
- •
- MS patients should undergo quantitative reevaluation of LVEF prior to each dose using the same methodology that was used to assess baseline LVEF. Additional doses of mitoxantrone should not be administered to multiple sclerosis patients who have experienced either a drop in LVEF to below the lower limit of normal or a clinically significant reduction in LVEF during mitoxantrone therapy.
- •
- MS patients should not receive a cumulative mitoxantrone dose greater than 140 mg/m2.
- •
- MS patients should undergo yearly quantitative LVEF evaluation after stopping mitoxantrone to monitor for late occurring cardiotoxicity.
For additional information, see WARNINGS and DOSAGE AND ADMINISTRATION.
Rx only
Mitoxantrone Injection Concentrate Description
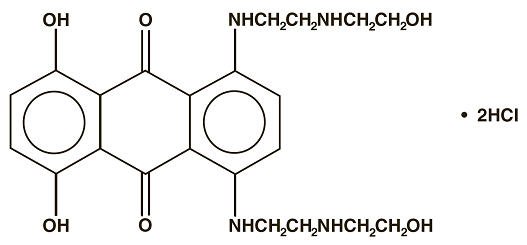
Mitoxantrone Injection, USP (concentrate) is a synthetic antineoplastic anthracenedione for intravenous use. The molecular formula is C22H28N4O6∙2HCl and the molecular weight is 517.41. It is supplied as a concentrate that MUST BE DILUTED PRIOR TO INJECTION. The concentrate is a sterile, nonpyrogenic, dark blue aqueous solution containing mitoxantrone hydrochloride equivalent to 2 mg/mL mitoxantrone free base, with sodium chloride (0.80% w/v), sodium metabisulfite (0.01% w/v), sodium acetate (0.005% w/v), acetic acid (0.046% w/v), and Water for Injection, USP as inactive ingredients. The solution has a pH of 3.0 to 4.5 and contains 0.14 mEq of sodium per mL. The product does not contain preservatives. The chemical name is 1, 4-dihydroxy-5, 8-bis[[2-[(2-hydroxyethyl) amino]ethyl]amino]-9,10- anthracenedione dihydrochloride and the structural formula is:
Mitoxantrone Injection Concentrate - Clinical Pharmacology
Mechanism of Action
Mitoxantrone, a DNA-reactive agent that intercalates into deoxyribonucleic acid (DNA) through hydrogen bonding, causes crosslinks and strand breaks. Mitoxantrone also interferes with ribonucleic acid (RNA) and is a potent inhibitor of topoisomerase II, an enzyme responsible for uncoiling and repairing damaged DNA. It has a cytocidal effect on both proliferating and nonproliferating cultured human cells, suggesting lack of cell cycle phase specificity.
Mitoxantrone has been shown in vitro to inhibit B cell, T cell, and macrophage proliferation and impair antigen presentation, as well as the secretion of interferon gamma, TNFα, and IL-2.
Pharmacokinetics
Pharmacokinetics of mitoxantrone in patients following a single intravenous administration of mitoxantrone can be characterized by a three-compartment model. The mean alpha half-life of mitoxantrone is 6 to 12 minutes, the mean beta half-life is 1.1 to 3.1 hours and the mean gamma (terminal or elimination) half-life is 23 to 215 hours (median approximately 75 hours). Pharmacokinetic studies have not been performed in humans receiving multiple daily dosing. Distribution to tissues is extensive: steady-state volume of distribution exceeds 1,000 L/m2. Tissue concentrations of mitoxantrone appear to exceed those in the blood during the terminal elimination phase. In the healthy monkey, distribution to brain, spinal cord, eye, and spinal fluid is low.
In patients administered 15 to 90 mg/m2 of mitoxantrone intravenously, there is a linear relationship between dose and the area under the concentration-time curve (AUC).
Mitoxantrone is 78% bound to plasma proteins in the observed concentration range of 26 to 455 ng/mL. This binding is independent of concentration and is not affected by the presence of phenytoin, doxorubicin, methotrexate, prednisone, prednisolone, heparin, or aspirin.
Metabolism and Elimination
Mitoxantrone is excreted in urine and feces as either unchanged drug or as inactive metabolites. In human studies, 11% and 25% of the dose were recovered in urine and feces, respectively, as either parent drug or metabolite during the 5-day period following drug administration. Of the material recovered in urine, 65% was unchanged drug. The remaining 35% was composed of monocarboxylic and dicarboxylic acid derivatives and their glucuronide conjugates. The pathways leading to the metabolism of mitoxantrone have not been elucidated.
Special Populations
Geriatric
In elderly patients with breast cancer, the systemic mitoxantrone clearance was 21.3 L/hr/m2, compared with 28.3 L/hr/m2 and 16.2 L/hr/m2 for non-elderly patients with nasopharyngeal carcinoma and malignant lymphoma, respectively.
Hepatic Impairment
Mitoxantrone clearance is reduced by hepatic impairment. Patients with severe hepatic dysfunction (bilirubin > 3.4 mg/dL) have an AUC more than three times greater than that of patients with normal hepatic function receiving the same dose. Patients with multiple sclerosis who have hepatic impairment should ordinarily not be treated with mitoxantrone. Other patients with hepatic impairment should be treated with caution and dosage adjustment may be required.
Drug Interactions
In vitro drug interaction studies have demonstrated that mitoxantrone did not inhibit CYP450 1A2, 2A6, 2C9, 2C19, 2D6, 2E1, and 3A4 across a broad concentration range. The results of in vitro induction studies are inconclusive, but suggest that mitoxantrone may be a weak inducer of CYP450 2E1 activity.
Pharmacokinetic studies of the interaction of mitoxantrone with concomitantly administered medications in humans have not been performed. The pathways leading to the metabolism of mitoxantrone have not been elucidated. To date, post-marketing experience has not revealed any significant drug interactions in patients who have received mitoxantrone for treatment of cancer. Information on drug interactions in patients with multiple sclerosis is limited.
Clinical Studies
Multiple Sclerosis
The safety and efficacy of mitoxantrone in multiple sclerosis were assessed in two randomized, multicenter clinical studies.
One randomized, controlled study (Study 1) was conducted in patients with secondary progressive or progressive relapsing multiple sclerosis. Patients in this study demonstrated significant neurological disability based on the Kurtzke Expanded Disability Status Scale (EDSS). The EDSS is an ordinal scale with 0.5 point increments ranging from 0.0 to 10.0 (increasing score indicates worsening) and based largely on ambulatory impairment in its middle range (EDSS 4.5 to 7.5 points). Patients in this study had experienced a mean deterioration in EDSS of about 1.6 points over the 18 months prior to enrollment.
Patients were randomized to receive placebo, 5 mg/m2 mitoxantrone, or 12 mg/m2 mitoxantrone administered IV every 3 months for 2 years. High-dose methylprednisolone was administered to treat relapses. The intent-to-treat analysis cohort consisted of 188 patients; 149 completed the 2-year study. Patients were evaluated every 3 months, and clinical outcome was determined after 24 months. In addition, a subset of patients was assessed with magnetic resonance imaging (MRI) at baseline, Month 12, and Month 24. Neurologic assessments and MRI reviews were performed by evaluators blinded to study drug and clinical outcome, although the diagnosis of relapse and the decision to treat relapses with steroids were made by unblinded treating physicians. A multivariate analysis of five clinical variables (EDSS, Ambulation Index [AI], number of relapses requiring treatment with steroids, months to first relapse needing treatment with steroids, and Standard Neurological Status [SNS]) was used to determine primary efficacy. The AI is an ordinal scale ranging from 0 to 9 in one point increments to define progressive ambulatory impairment. The SNS provides an overall measure of neurologic impairment and disability, with scores ranging from 0 (normal neurologic examination) to 99 (worst possible score).
Results of Study 1 are summarized in Table 1.
| Treatment Groups | p-value | |||
|---|---|---|---|---|
| Mitoxantrone | Placebo vs. | |||
| Primary Endpoints | Placebo
(N = 64) | 5 mg/m2
(N = 64) | 12 mg/m2
(N = 60) | 12 mg/m2
Mitoxantrone |
| NR = not reached within 24 months; MRI = magnetic resonance imaging. | ||||
|
Primary efficacy multivariate analysis* |
- |
- |
- |
< 0.0001 |
|
Primary clinical variables analyzed: |
||||
|
EDSS change† (mean) |
0.23 |
– 0.23 |
– 0.13 |
0.0194 |
|
Ambulation Index change† (mean) |
0.77 |
0.41 |
0.30 |
0.0306 |
|
Mean number of relapses per patient requiring corticosteroid treatment (adjusted for discontinuation) |
1.20 |
0.73 |
0.40 |
0.0002 |
|
Months to first relapse requiring corticosteroid treatment (median [1st quartile]) |
14.2 [6.7] |
NR [6.9] |
NR [20.4] |
0.0004 |
|
Standard Neurological Status change† (mean) |
0.77 |
– 0.38 |
– 1.07 |
0.0269 |
|
MRI‡ |
||||
|
No. of patients with new Gd-enhancing lesions |
5/32 (16%) |
4/37 (11%) |
0/31 |
0.022 |
|
Change in number of T2-weighted lesions, mean (n)† |
1.94 (32) |
0.68 (34) |
0.29 (28) |
0.027 |
A second randomized, controlled study (Study 2) evaluated mitoxantrone in combination with methylprednisolone (MP) and was conducted in patients with secondary progressive or worsening relapsing-remitting multiple sclerosis who had residual neurological deficit between relapses. All patients had experienced at least two relapses with sequelae or neurological deterioration within the previous 12 months. The average deterioration in EDSS was 2.2 points during the previous 12 months. During the screening period, patients were treated with two monthly doses of 1 g of IV MP and underwent monthly MRI scans. Only patients who developed at least one new Gd-enhancing MRI lesion during the 2-month screening period were eligible for randomization. A total of 42 evaluable patients received monthly treatments of 1 g of IV MP alone (n = 21) or ~12 mg/m2 of IV mitoxantrone plus 1 g of IV MP (n = 21) (MIT + MP) for 6 months. Patients were evaluated monthly, and study outcome was determined after 6 months. The primary measure of effectiveness in this study was a comparison of the proportion of patients in each treatment group who developed no new Gd-enhancing MRI lesions at 6 months; these MRIs were assessed by a blinded panel. Additional outcomes were measured, including EDSS and number of relapses, but all clinical measures in this trial were assessed by an unblinded treating physician. Five patients, all in the MP alone arm, failed to complete the study due to lack of efficacy.
The results of this trial are displayed in Table 2.
| Primary Endpoint | MP alone
(N = 21) | MIT + MP
(N = 21) | p-value |
|---|---|---|---|
| MP = methylprednisolone; MIT + MP = mitoxantrone plus methylprednisolone. | |||
|
|||
|
Patients (%) without new Gd-enhancing lesions on MRIs (primary endpoint)* |
5 (31%) |
19 (90%) |
0.001 |
|
Secondary Endpoints |
|||
|
EDSS change (Month 6 minus baseline)* (mean) |
– 0.1 |
– 1.1 |
0.013 |
|
Annualized relapse rate (mean per patient) |
3.0 |
0.7 |
0.003 |
|
Patients (%) without relapses |
7 (33%) |
14 (67%) |
0.031 |
Advanced Hormone-Refractory Prostate Cancer
A multicenter Phase 2 trial of mitoxantrone and low-dose prednisone (M + P) was conducted in 27 symptomatic patients with hormone-refractory prostate cancer. Using NPCP (National Prostate Cancer Project) criteria for disease response, there was one partial responder and 12 patients with stable disease. However, nine patients or 33% achieved a palliative response defined on the basis of reduction in analgesic use or pain intensity.
These findings led to the initiation of a randomized multicenter trial (CCI-NOV22) comparing the effectiveness of (M + P) to low-dose prednisone alone (P). Eligible patients were required to have metastatic or locally advanced disease that had progressed on standard hormonal therapy, a castrate serum testosterone level, and at least mild pain at study entry. Mitoxantrone was administered at a dose of 12 mg/m2 by short IV infusion every 3 weeks. Prednisone was administered orally at a dose of 5 mg twice a day. Patients randomized to the prednisone arm were crossed over to the M + P arm if they progressed or if they were not improved after a minimum of 6 weeks of therapy with prednisone alone.
A total of 161 patients were randomized, 80 to the M + P arm and 81 to the P arm. The median mitoxantrone dose administered was 12 mg/m2 per cycle. The median cumulative mitoxantrone dose administered was 73 mg/m2 (range of 12 to 212 mg/m2).
A primary palliative response (defined as a 2-point decrease in pain intensity in a 6-point pain scale, associated with stable analgesic use, and lasting a minimum of 6 weeks) was achieved in 29% of patients randomized to M + P compared to 12% of patients randomized to P alone (p = 0.011). Two responders left the study after meeting primary response criterion for two consecutive cycles. For the purposes of this analysis, these two patients were assigned a response duration of zero days. A secondary palliative response was defined as a 50% or greater decrease in analgesic use, associated with stable pain intensity, and lasting a minimum of 6 weeks. An overall palliative response (defined as primary plus secondary responses) was achieved in 38% of patients randomized to M + P compared to 21% of patients randomized to P (p = 0.025).
The median duration of primary palliative response for patients randomized to M + P was 7.6 months compared to 2.1 months for patients randomized to P alone (p = 0.0009). The median duration of overall palliative response for patients randomized to M + P was 5.6 months compared to 1.9 months for patients randomized to P alone (p = 0.0004).
Time to progression was defined as a 1-point increase in pain intensity, or a > 25% increase in analgesic use, or evidence of disease progression on radiographic studies, or requirement for radiotherapy. The median time to progression for all patients randomized to M + P was 4.4 months compared to 2.3 months for all patients randomized to P alone (p = 0.0001). Median time to death was 11.3 months for all patients on the M + P arm compared to 10.8 months for all patients on P alone (p = 0.2324).
Forty-eight patients on the P arm crossed over to receive M + P. Of these, thirty patients had progressed on P, while 18 had stable disease on P. The median cycle of crossover was 5 cycles (range of 2 to 16 cycles). Time trends for pain intensity prior to crossover were significantly worse for patients who crossed over than for those who remained on P alone (p = 0.012). Nine patients (19%) demonstrated a palliative response on M + P after crossover. The median time to death for patients who crossed over to M + P was 12.7 months.
The clinical significance of a fall in prostate-specific antigen (PSA) concentrations after chemotherapy is unclear. On the CCI-NOV22 trial, a PSA fall of 50% or greater for two consecutive follow-up assessments after baseline was reported in 33% of all patients randomized to the M + P arm and 9% of all patients randomized to the P arm. These findings should be interpreted with caution since PSA responses were not defined prospectively. A number of patients were inevaluable for response, and there was an imbalance between treatment arms in the numbers of evaluable patients. In addition, PSA reduction did not correlate precisely with palliative response, the primary efficacy endpoint of this study. For example, among the 26 evaluable patients randomized to the M + P arm who had ≥ 50% reduction in PSA, only 13 had a primary palliative response. Also, among 42 evaluable patients on this arm who did not have this reduction in PSA, 8 nonetheless had a primary palliative response.
Investigators at Cancer and Leukemia Group B (CALGB) conducted a Phase 3 comparative trial of mitoxantrone plus hydrocortisone (M + H) versus hydrocortisone alone (H) in patients with hormone-refractory prostate cancer (CALGB 9182). Eligible patients were required to have metastatic disease that had progressed despite at least one hormonal therapy. Progression at study entry was defined on the basis of progressive symptoms, increases in measurable or osseous disease, or rising PSA levels. Mitoxantrone was administered intravenously at a dose of 14 mg/m2 every 21 days and hydrocortisone was administered orally at a daily dose of 40 mg. A total of 242 subjects were randomized, 119 to the M + H arm and 123 to the H arm. There were no differences in survival between the two arms, with a median of 11.1 months in the M + H arm and 12 months in the H arm (p = 0.3298).
Using NPCP criteria for response, partial responses were achieved in 10 patients (8.4%) randomized to the M + H arm compared with 2 patients (1.6%) randomized to the H arm (p = 0.018). The median time to progression, defined by NPCP criteria, for patients randomized to the M + H arm was 7.3 months compared to 4.1 months for patients randomized to H alone (p = 0.0654).
Approximately 60% of patients on each arm required analgesics at baseline. Analgesic use was measured in this study using a 5-point scale. The best percent change from baseline in mean analgesic use was -17% for 61 patients with available data on the M + H arm, compared with +17% for 61 patients on H alone (p = 0.014). A time trend analysis for analgesic use in individual patients also showed a trend favoring the M + H arm over H alone but was not statistically significant.
Pain intensity was measured using the Symptom Distress Scale (SDS) Pain Item 2 (a 5-point scale). The best percent change from baseline in mean pain intensity was -14% for 37 patients with available data on the M + H arm, compared with +8% for 38 patients on H alone (p = 0.057). A time trend analysis for pain intensity in individual patients showed no difference between treatment arms.
Acute Nonlymphocytic Leukemia
In two large randomized multicenter trials, remission induction therapy for acute nonlymphocytic leukemia (ANLL) with mitoxantrone 12 mg/m2 daily for 3 days as a 10-minute intravenous infusion and cytarabine 100 mg/m2 for 7 days given as a continuous 24-hour infusion was compared with daunorubicin 45 mg/m2 daily by intravenous infusion for 3 days plus the same dose and schedule of cytarabine used with mitoxantrone. Patients who had an incomplete antileukemic response received a second induction course in which mitoxantrone or daunorubicin was administered for 2 days and cytarabine for 5 days using the same daily dosage schedule. Response rates and median survival information for both the U.S. and international multicenter trials are given in Table 3:
| Trial | % Complete Response (CR) | Median Time to CR (days) | Survival (days) | |||
|---|---|---|---|---|---|---|
| MIT | DAUN | MIT | DAUN | MIT | DAUN | |
| MIT = mitoxantrone + cytarabine DAUN = daunorubicin + cytarabine |
||||||
|
U.S. |
63 (62/98) |
53 (54/102) |
35 |
42 |
312 |
237 |
|
International |
50 (56/112) |
51 (62/123) |
36 |
42 |
192 |
230 |
In these studies, two consolidation courses were administered to complete responders on each arm. Consolidation therapy consisted of the same drug and daily dosage used for remission induction, but only 5 days of cytarabine and 2 days of mitoxantrone or daunorubicin were given. The first consolidation course was administered 6 weeks after the start of the final induction course if the patient achieved a complete remission. The second consolidation course was generally administered 4 weeks later. Full hematologic recovery was necessary for patients to receive consolidation therapy. For the U.S. trial, median granulocyte nadirs for patients receiving mitoxantrone + cytarabine for consolidation courses 1 and 2 were 10/mm3 for both courses, and for those patients receiving daunorubicin + cytarabine nadirs were 170/mm3 and 260/mm3, respectively. Median platelet nadirs for patients who received mitoxantrone + cytarabine for consolidation courses 1 and 2 were 17,000/mm3 and 14,000/mm3, respectively, and were 33,000/mm3 and 22,000/mm3 in courses 1 and 2 for those patients who received daunorubicin + cytarabine. The benefit of consolidation therapy in ANLL patients who achieve a complete remission remains controversial. However, in the only well-controlled prospective, randomized multicenter trials with mitoxantrone in ANLL, consolidation therapy was given to all patients who achieved a complete remission. During consolidation in the U.S. study, two myelosuppression-related deaths occurred in the mitoxantrone arm and one on the daunorubicin arm. However, in the international study there were eight deaths on the mitoxantrone arm during consolidation which were related to the myelosuppression and none on the daunorubicin arm where less myelosuppression occurred.
Indications and Usage for Mitoxantrone Injection Concentrate
Mitoxantrone is indicated for reducing neurologic disability and/or the frequency of clinical relapses in patients with secondary (chronic) progressive, progressive relapsing, or worsening relapsing-remitting multiple sclerosis (i.e., patients whose neurologic status is significantly abnormal between relapses). Mitoxantrone is not indicated in the treatment of patients with primary progressive multiple sclerosis.
The clinical patterns of multiple sclerosis in the studies were characterized as follows: secondary progressive and progressive relapsing disease were characterized by gradual increasing disability with or without superimposed clinical relapses, and worsening relapsing-remitting disease was characterized by clinical relapses resulting in a step-wise worsening of disability.
Mitoxantrone in combination with corticosteroids is indicated as initial chemotherapy for the treatment of patients with pain related to advanced hormone-refractory prostate cancer.
Mitoxantrone in combination with other approved drug(s) is indicated in the initial therapy of acute nonlymphocytic leukemia (ANLL) in adults. This category includes myelogenous, promyelocytic, monocytic, and erythroid acute leukemias.
Contraindications
Mitoxantrone is contraindicated in patients who have demonstrated prior hypersensitivity to it.
Warnings
WHEN MITOXANTRONE IS USED IN HIGH DOSES (> 14 mg/m2/d × 3 days) SUCH AS INDICATED FOR THE TREATMENT OF LEUKEMIA, SEVERE MYELOSUPPRESSION WILL OCCUR. THEREFORE, IT IS RECOMMENDED THAT MITOXANTRONE BE ADMINISTERED ONLY BY PHYSICIANS EXPERIENCED IN THE CHEMOTHERAPY OF THIS DISEASE. LABORATORY AND SUPPORTIVE SERVICES MUST BE AVAILABLE FOR HEMATOLOGIC AND CHEMISTRY MONITORING AND ADJUNCTIVE THERAPIES, INCLUDING ANTIBIOTICS.
BLOOD AND BLOOD PRODUCTS MUST BE AVAILABLE TO SUPPORT PATIENTS DURING THE EXPECTED PERIOD OF MEDULLARY HYPOPLASIA AND SEVERE MYELOSUPPRESSION. PARTICULAR CARE SHOULD BE GIVEN TO ASSURING FULL HEMATOLOGIC RECOVERY BEFORE UNDERTAKING CONSOLIDATION THERAPY (IF THIS TREATMENT IS USED) AND PATIENTS SHOULD BE MONITORED CLOSELY DURING THIS PHASE. MITOXANTRONE ADMINISTERED AT ANY DOSE CAN CAUSE MYELOSUPPRESSION.
CONTAINS SODIUM METABISULFITE, A SULFITE THAT MAY CAUSE ALLERGIC-TYPE REACTIONS INCLUDING ANAPHYLACTIC SYMPTOMS AND LIFE-THREATENING OR LESS SEVERE ASTHMATIC EPISODES IN CERTAIN SUSCEPTIBLE PEOPLE. THE OVERALL PREVALENCE OF SULFITE SENSITIVITY IN THE GENERAL POPULATION IS UNKNOWN AND PROBABLY LOW. SULFITE SENSITIVITY IS SEEN MORE FREQUENTLY IN ASTHMATIC THAN IN NONASTHMATIC PEOPLE.
General
Patients with preexisting myelosuppression as the result of prior drug therapy should not receive mitoxantrone unless it is felt that the possible benefit from such treatment warrants the risk of further medullary suppression.
The safety of Mitoxantrone Injection, USP (concentrate) in patients with hepatic insufficiency is not established (see CLINICAL PHARMACOLOGY).
Safety for use by routes other than intravenous administration has not been established.
Mitoxantrone is not indicated for subcutaneous, intramuscular, or intra-arterial injection. There have been reports of local/regional neuropathy, some irreversible, following intra-arterial injection.
Mitoxantrone must not be given by intrathecal injection. There have been reports of neuropathy and neurotoxicity, both central and peripheral, following intrathecal injection. These reports have included seizures leading to coma and severe neurologic sequelae, and paralysis with bowel and bladder dysfunction.
Topoisomerase II inhibitors, including mitoxantrone, have been associated with the development of secondary acute myeloid leukemia and myelosuppression.
Cardiac Effects
Because of the possible danger of cardiac effects in patients previously treated with daunorubicin or doxorubicin, the benefit-to-risk ratio of mitoxantrone therapy in such patients should be determined before starting therapy.
Functional cardiac changes including decreases in left ventricular ejection fraction (LVEF) and irreversible congestive heart failure can occur with mitoxantrone. Cardiac toxicity may be more common in patients with prior treatment with anthracyclines, prior mediastinal radiotherapy, or with preexisting cardiovascular disease. Such patients should have regular cardiac monitoring of LVEF from the initiation of therapy. Cancer patients who received cumulative doses of 140 mg/m2 either alone or in combination with other chemotherapeutic agents had a cumulative 2.6% probability of clinical congestive heart failure. In comparative oncology trials, the overall cumulative probability rate of moderate or severe decreases in LVEF at this dose was 13%.
Multiple Sclerosis
Changes in cardiac function may occur in patients with multiple sclerosis treated with mitoxantrone. In one controlled trial (Study 1, see CLINICAL TRIALS, Multiple Sclerosis), two patients (2%) of 127 receiving mitoxantrone, one receiving a 5 mg/m2 dose and the other receiving the 12 mg/m2 dose, had LVEF values that decreased to below 50%. An additional patient receiving 12 mg/m2, who did not have LVEF measured, had a decrease in another echocardiographic measurement of ventricular function (fractional shortening) that led to discontinuation from the trial (see ADVERSE REACTIONS, Multiple Sclerosis). There were no reports of congestive heart failure in either controlled trial.
MS patients should be assessed for cardiac signs and symptoms by history, physical examination, ECG, and quantitative LVEF evaluation using appropriate methodology (ex. Echocardiogram, MUGA, MRI, etc.) prior to the start of mitoxantrone therapy. MS patients with a baseline LVEF below the lower limit of normal should not be treated with mitoxantrone. Subsequent LVEF and ECG evaluations are recommended if signs or symptoms of congestive heart failure develop and prior to every dose administered to MS patients. Mitoxantrone should not be administered to MS patients who experience a reduction in LVEF to below the lower limit of normal, to those who experience a clinically significant reduction in LVEF, or to those who have received a cumulative lifetime dose of 140 mg/m2. MS patients should have yearly quantitative LVEF evaluation after stopping mitoxantrone to monitor for late-occurring cardiotoxicity.
Leukemia
Acute congestive heart failure may occasionally occur in patients treated with mitoxantrone for ANLL. In first-line comparative trials of mitoxantrone + cytarabine vs daunorubicin + cytarabine in adult patients with previously untreated ANLL, therapy was associated with congestive heart failure in 6.5% of patients on each arm. A causal relationship between drug therapy and cardiac effects is difficult to establish in this setting since myocardial function is frequently depressed by the anemia, fever and infection, and hemorrhage that often accompany the underlying disease.
Hormone-Refractory Prostate Cancer
Functional cardiac changes such as decreases in LVEF and congestive heart failure may occur in patients with hormone-refractory prostate cancer treated with mitoxantrone. In a randomized comparative trial of mitoxantrone plus low-dose prednisone vs low-dose prednisone, 7 of 128 patients (5.5%) treated with mitoxantrone had a cardiac event defined as any decrease in LVEF below the normal range, congestive heart failure (n = 3), or myocardial ischemia. Two patients had a prior history of cardiac disease. The total mitoxantrone dose administered to patients with cardiac effects ranged from > 48 to 212 mg/m2.
Among 112 patients evaluable for safety on the mitoxantrone + hydrocortisone arm of the CALGB trial, 18 patients (19%) had a reduction in cardiac function, 5 patients (5%) had cardiac ischemia, and 2 patients (2%) experienced pulmonary edema. The range of total mitoxantrone doses administered to these patients is not available.
Pregnancy
Mitoxantrone may cause fetal harm when administered to a pregnant woman. Women of childbearing potential should be advised to avoid becoming pregnant. Mitoxantrone is considered a potential human teratogen because of its mechanism of action and the developmental effects demonstrated by related agents. Treatment of pregnant rats during the organogenesis period of gestation was associated with fetal growth retardation at doses ≥0.1 mg/kg/day (0.01 times the recommended human dose on a mg/m2 basis). When pregnant rabbits were treated during organogenesis, an increased incidence of premature delivery was observed at doses ≥0.1 mg/kg/day (0.01 times the recommended human dose on a mg/m2 basis). No teratogenic effects were observed in these studies, but the maximum doses tested were well below the recommended human dose (0.02 and 0.05 times in rats and rabbits, respectively, on a mg/m2 basis). There are no adequate and well-controlled studies in pregnant women. Women with multiple sclerosis who are biologically capable of becoming pregnant should have a pregnancy test prior to each dose, and the results should be known prior to administration of the drug. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential risk to the fetus.
Secondary Leukemia
Mitoxantrone therapy increases the risk of developing secondary leukemia in patients with cancer and in patients with multiple sclerosis.
In a study of patients with prostate cancer, acute myeloid leukemia occurred in 1% (5/487) of mitoxantrone-treated patients versus no cases in the control group (0/496) not receiving mitoxantrone at 4.7 years followup.
In a prospective, open-label, tolerability and safety monitoring study of mitoxantrone treated MS patients followed for up to five years (median of 2.8 years), leukemia occurred in 0.6% (3/509) of patients. Publications describe leukemia risk of 0.25% to 2.8% in cohorts of patients with MS treated with mitoxantrone and followed for varying periods of time. This leukemia risk exceeds the risk of leukemia in the general population. The most commonly reported types were acute promyelocytic leukemia and acute myelocytic leukemia.
In 1774 patients with breast cancer who received mitoxantrone concomitantly with other cytotoxic agents and radiotherapy, the cumulative risk of developing treatment-related acute myeloid leukemia was estimated as 1.1% and 1.6% at 5 and 10 years, respectively. The second largest report involved 449 patients with breast cancer treated with mitoxantrone, usually in combination with radiotherapy and/or other cytotoxic agents. In this study, the cumulative probability of developing secondary leukemia was estimated to be 2.2% at 4 years.
Secondary acute myeloid leukemia has also been reported in cancer patients treated with anthracyclines. Mitoxantrone is an anthracenedione, a related drug. The occurrence of secondary leukemia is more common when anthracyclines are given in combination with DNA-damaging antineoplastic agents, when patients have been heavily pretreated with cytotoxic drugs, or when doses of anthracyclines have been escalated.
Symptoms of acute leukemia may include excessive bruising, bleeding, and recurrent infections.
Precautions
General
Therapy with mitoxantrone should be accompanied by close and frequent monitoring of hematologic and chemical laboratory parameters, as well as frequent patient observation.
Systemic infections should be treated concomitantly with or just prior to commencing therapy with mitoxantrone.
Information for Patients
See FDA-approved patient labeling (MEDICATION GUIDE).
Inform patients of the availability of a Medication Guide and instruct them to read the Medication Guide prior to initiating treatment with MitoXANTRONE and prior to each infusion. Review the MitoXANTRONE Medication Guide with every patient prior to initiation of treatment and periodically during treatment. Instruct patients that MitoXANTRONE should be taken only as prescribed.
Advise patients that MitoXANTRONE can cause myelosuppression and inform patients of the signs and symptoms of myelosuppression. Advise patients that MitoXANTRONE can cause congestive heart failure that may lead to death even in people who have never had heart problems before, and inform patients of the signs and symptoms of congestive heart failure. Advise patients receiving MitoXANTRONE to treat multiple sclerosis that they should receive cardiac monitoring prior to each MitoXANTRONE dose and yearly after stopping MitoXANTRONE.
MitoXANTRONE may impart a blue-green color to the urine for 24 hours after administration, and patients should be advised to expect this during therapy. Bluish discoloration of the sclera may also occur.
Laboratory Tests
A complete blood count, including platelets, should be obtained prior to each course of mitoxantrone and in the event that signs and symptoms of infection develop. Liver function tests should also be performed prior to each course of therapy. Mitoxantrone therapy in multiple sclerosis patients with abnormal liver function tests is not recommended because mitoxantrone clearance is reduced by hepatic impairment and no laboratory measurement can predict drug clearance and dose adjustments.
In leukemia treatment, hyperuricemia may occur as a result of rapid lysis of tumor cells by mitoxantrone. Serum uric acid levels should be monitored and hypouricemic therapy instituted prior to the initiation of antileukemic therapy.
Women with multiple sclerosis who are biologically capable of becoming pregnant, even if they are using birth control, should have a pregnancy test, and the results should be known, before receiving each dose of mitoxantrone (see WARNINGS, Pregnancy).
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Intravenous treatment of rats and mice, once every 21 days for 24 months, with mitoxantrone resulted in an increased incidence of fibroma and external auditory canal tumors in rats at a dose of 0.03 mg/kg (0.02 fold the recommended human dose, on a mg/m2 basis), and hepatocellular adenoma in male mice at a dose of 0.1 mg/kg (0.03 fold the recommended human dose, on a mg/m2 basis). Intravenous treatment of rats, once every 21 days for 12 months with mitoxantrone resulted in an increased incidence of external auditory canal tumors in rats at a dose of 0.3 mg/kg (0.15 fold the recommended human dose, on a mg/m2 basis).
Mutagenesis
Mitoxantrone was clastogenic in the in vivo rat bone marrow assay. Mitoxantrone was also clastogenic in two in vitro assays; it induced DNA damage in primary rat hepatocytes and sister chromatid exchanges in Chinese hamster ovary cells. Mitoxantrone was mutagenic in bacterial and mammalian test systems (Ames/Salmonella and E. coli and L5178Y TK+/-mouse lymphoma).
Drug Interactions
Mitoxantrone and its metabolites are excreted in bile and urine, but it is not known whether the metabolic or excretory pathways are saturable, may be inhibited or induced, or if mitoxantrone and its metabolites undergo enterohepatic circulation. To date, post-marketing experience has not revealed any significant drug interactions in patients who have received mitoxantrone for treatment of cancer. Information on drug interactions in patients with multiple sclerosis is limited.
Following concurrent administration of mitoxantrone with corticosteroids, no evidence of drug interactions has been observed.
Special Populations
Hepatic Impairment
Patients with multiple sclerosis who have hepatic impairment should ordinarily not be treated with mitoxantrone. Mitoxantrone should be administered with caution to other patients with hepatic impairment. In patients with severe hepatic impairment, the AUC is more than three times greater than the value observed in patients with normal hepatic function.
Nursing Mothers
Mitoxantrone is excreted in human milk and significant concentrations (18 ng/mL) have been reported for 28 days after the last administration. Because of the potential for serious adverse reactions in infants from mitoxantrone, breast feeding should be discontinued before starting treatment.
Geriatric Use
Multiple Sclerosis
Clinical studies of mitoxantrone did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
Hormone-Refractory Prostate Cancer
One hundred forty-six patients aged 65 and over and 52 younger patients (<65 years) have been treated with mitoxantrone in controlled clinical studies. These studies did not include sufficient numbers of younger patients to determine whether they respond differently from older patients. However, greater sensitivity of some older individuals cannot be ruled out.
Adverse Reactions/Side Effects
Multiple Sclerosis
Mitoxantrone has been administered to 149 patients with multiple sclerosis in two randomized clinical trials, including 21 patients who received mitoxantrone in combination with corticosteroids.
In Study 1, the proportion of patients who discontinued treatment due to an adverse event was 9.7% (n = 6) in the 12 mg/m2 mitoxantrone arm (leukopenia, depression, decreased LV function, bone pain and emesis, renal failure, and one discontinuation to prevent future complications from repeated urinary tract infections) compared to 3.1% (n = 2) in the placebo arm (hepatitis and myocardial infarction). The following clinical adverse experiences were significantly more frequent in the mitoxantrone groups: nausea, alopecia, urinary tract infection, and menstrual disorders, including amenorrhea.
Table 4a summarizes clinical adverse events of all intensities occurring in ≥5% of patients in either dose group of mitoxantrone and that were numerically greater on drug than on placebo in Study 1. The majority of these events were of mild to moderate intensity, and nausea was the only adverse event that occurred with severe intensity in more than one patient (three patients [5%] in the 12 mg/m2 group). Of note, alopecia consisted of mild hair thinning.
Two of the 127 patients treated with mitoxantrone in Study 1 had decreased LVEF to below 50% at some point during the 2 years of treatment. An additional patient receiving 12 mg/m2 did not have LVEF measured, but had another echocardiographic measure of ventricular function (fractional shortening) that led to discontinuation from the study.
| Percent of Patients | |||
|---|---|---|---|
| Preferred Term | Placebo (N = 64) | 5 mg/m2
Mitoxantrone (N = 65) | 12 mg/m2
Mitoxantrone (N = 62) |
|
|||
|
Nausea |
20 |
55 |
76 |
|
Alopecia |
31 |
38 |
61 |
|
Menstrual disorder* |
26 |
51 |
61 |
|
Amenorrhea* |
3 |
28 |
43 |
|
Upper respiratory tract infection |
52 |
51 |
53 |
|
Urinary tract infection |
13 |
29 |
32 |
|
Stomatitis |
8 |
15 |
19 |
|
Arrhythmia |
8 |
6 |
18 |
|
Diarrhea |
11 |
25 |
16 |
|
Urine abnormal |
6 |
5 |
11 |
|
ECG abnormal |
3 |
5 |
11 |
|
Constipation |
6 |
14 |
10 |
|
Back pain |
5 |
6 |
8 |
|
Sinusitis |
2 |
3 |
6 |
|
Headache |
5 |
6 |
6 |
The proportion of patients experiencing any infection during Study 1 was 67% for the placebo group, 85% for the 5 mg/m2 group, and 81% for the 12 mg/m2 group. However, few of these infections required hospitalization: one placebo patient (tonsillitis), three 5 mg/m2 patients (enteritis, urinary tract infection, viral infection), and four 12 mg/m2 patients (tonsillitis, urinary tract infection [two], endometritis).
Table 4b summarizes laboratory abnormalities that occurred in ≥ 5% of patients in either mitoxantrone dose group, and that were numerically more frequent than in the placebo group.
| Percent of Patients | |||
|---|---|---|---|
| Event | Placebo (N = 64) | 5 mg/m2
Mitoxantrone (N = 65) | 12 mg/m2
Mitoxantrone (N = 62) |
|
Leukopenia† |
0 |
9 |
19 |
|
Gamma-GT increased |
3 |
3 |
15 |
|
SGOT increased |
8 |
9 |
8 |
|
Granulocytopenia‡ |
2 |
6 |
6 |
|
Anemia |
2 |
9 |
6 |
|
SGPT increased |
3 |
6 |
5 |
There was no difference among treatment groups in the incidence or severity of hemorrhagic events.
In Study 2, mitoxantrone was administered once a month. Clinical adverse events most frequently reported in the mitoxantrone group included amenorrhea (53% of female patients), alopecia (33% of patients), nausea (29% of patients), and asthenia (24% of patients). Tables 5a and 5b respectively summarize adverse events and laboratory abnormalities occurring in > 5% of patients in the mitoxantrone group and numerically more frequent than in the control group.
| Percent of Patients | ||
|---|---|---|
| Event | MP (N = 21) | M + MP (N = 21) |
| M = mitoxantrone, MP = methylprednisolone | ||
|
Amenorrhea† |
0 |
53 |
|
Alopecia |
0 |
33 |
|
Nausea |
0 |
29 |
|
Asthenia |
0 |
24 |
|
Pharyngitis/throat infection |
5 |
19 |
|
Gastralgia/stomach burn/epigastric pain |
5 |
14 |
|
Aphthosis |
0 |
10 |
|
Cutaneous mycosis |
0 |
10 |
|
Rhinitis |
0 |
10 |
|
Menorrhagia† |
0 |
7 |
| Percent of Patients | ||
|---|---|---|
| Event | MP (N = 21) | M + MP (N = 21) |
| M = mitoxantrone, MP = methylprednisolone | ||
|
WBC low† |
14 |
100 |
|
ANC low‡ |
10 |
100 |
|
Lymphocytes low |
43 |
95 |
|
Hemoglobin low |
48 |
43 |
|
Platelets low§ |
0 |
33 |
|
SGOT high |
5 |
15 |
|
SGPT high |
10 |
15 |
|
Glucose high |
5 |
10 |
|
Potassium low |
0 |
10 |
Leukopenia and neutropenia were reported in the M + MP group (see Table 5b).
Neutropenia occurred within 3 weeks after mitoxantrone administration and was always reversible. Only mild to moderate intensity infections were reported in 9 of 21 patients in the M + MP group and in 3 of 21 patients in the MP group; none of these required hospitalization. There was no difference among treatment groups in the incidence or severity of hemorrhagic events. There were no withdrawals from Study 2 for safety reasons.
Leukemia
Mitoxantrone has been studied in approximately 600 patients with acute non-lymphocytic leukemia (ANLL). Table 6 represents the adverse reaction experience in the large U.S. comparative study of mitoxantrone + cytarabine vs daunorubicin + cytarabine. Experience in the large international study was similar. A much wider experience in a variety of other tumor types revealed no additional important reactions other than cardiomyopathy (see WARNINGS). It should be appreciated that the listed adverse reaction categories include overlapping clinical symptoms related to the same condition, e.g., dyspnea, cough and pneumonia. In addition, the listed adverse reactions cannot all necessarily be attributed to chemotherapy as it is often impossible to distinguish effects of the drug and effects of the underlying disease. It is clear, however, that the combination of mitoxantrone + cytarabine was responsible for nausea and vomiting, alopecia, mucositis/stomatitis, and myelosuppression.
Table 6 summarizes adverse reactions occurring in patients treated with mitoxantrone + cytarabine in comparison with those who received daunorubicin + cytarabine for therapy of ANLL in a large multicenter randomized prospective U.S. trial.
Adverse reactions are presented as major categories and selected examples of clinically significant subcategories.
| Induction
[% pts entering induction] | Consolidation
[% pts entering induction] |
|||
|---|---|---|---|---|
| Event | MIT
N = 102 | DAUN
N = 102 | MIT
N = 55 | DAUN
N = 49 |
| MIT = mitoxantrone, DAUN = daunorubicin. | ||||
|
Cardiovascular |
26 |
28 |
11 |
24 |
|
CHF |
5 |
6 |
0 |
0 |
|
Arrhythmias |
3 |
3 |
4 |
4 |
|
Bleeding |
37 |
41 |
20 |
6 |
|
GI |
16 |
12 |
2 |
2 |
|
Petechiae/ecchymoses |
7 |
9 |
11 |
2 |
|
Gastrointestinal |
88 |
85 |
58 |
51 |
|
Nausea/vomiting |
72 |
67 |
31 |
31 |
|
Diarrhea |
47 |
47 |
18 |
8 |
|
Abdominal pain |
15 |
9 |
9 |
4 |
|
Mucositis/stomatitis |
29 |
33 |
18 |
8 |
|
Hepatic |
10 |
11 |
14 |
2 |
|
Jaundice |
3 |
8 |
7 |
0 |
|
Infections |
66 |
73 |
60 |
43 |
|
UTI |
7 |
2 |
7 |
2 |
|
Pneumonia |
9 |
7 |
9 |
0 |
|
Sepsis |
34 |
36 |
31 |
18 |
|
Fungal infections |
15 |
13 |
9 |
6 |
|
Renal failure |
8 |
6 |
0 |
2 |
|
Fever |
78 |
71 |
24 |
18 |
|
Alopecia |
37 |
40 |
22 |
16 |
|
Pulmonary |
43 |
43 |
24 |
14 |
|
Cough |
13 |
9 |
9 |
2 |
|
Dyspnea |
18 |
20 |
6 |
0 |
|
CNS |
30 |
30 |
34 |
35 |
|
Seizures |
4 |
4 |
2 |
8 |
|
Headache |
10 |
9 |
13 |
8 |
|
Eye |
7 |
6 |
2 |
4 |
|
Conjunctivitis |
5 |
1 |
0 |
0 |
Hormone-Refractory Prostate Cancer
Detailed safety information is available for a total of 353 patients with hormone-refractory prostate cancer treated with mitoxantrone, including 274 patients who received mitoxantrone in combination with corticosteroids.
Table 7 summarizes adverse reactions of all grades occurring in ≥5% of patients in Trial CCI-NOV22.
| Event | M + P (n = 80) % | P (n = 81) % |
|---|---|---|
| M = mitoxantrone, P = prednisone. No nonhematologic adverse events of Grade 3/4 were seen in > 5% of patients. |
||
|
Nausea |
61 |
35 |
|
Fatigue |
39 |
14 |
|
Alopecia |
29 |
0 |
|
Anorexia |
25 |
6 |
|
Constipation |
16 |
14 |
|
Dyspnea |
11 |
5 |
|
Nail bed changes |
11 |
0 |
|
Edema |
10 |
4 |
|
Systemic infection |
10 |
7 |
|
Mucositis |
10 |
0 |
|
UTI |
9 |
4 |
|
Emesis |
9 |
5 |
|
Pain |
8 |
9 |
|
Fever |
6 |
3 |
|
Hemorrhage/bruise |
6 |
1 |
|
Anemia |
5 |
3 |
|
Cough |
5 |
0 |
|
Decreased LVEF |
5 |
0 |
|
Anxiety/depression |
5 |
3 |
|
Dyspepsia |
5 |
6 |
|
Skin infection |
5 |
3 |
|
Blurred vision |
3 |
5 |
Table 8 summarizes adverse events of all grades occurring in ≥ 5% of patients in Trial CALGB 9182.
| M + H (n = 112) | H (n = 113) | |||
|---|---|---|---|---|
| Event | n | % | n | % |
| M = mitoxantrone, H = hydrocortisone | ||||
|
Decreased WBC |
96 |
87 |
4 |
4 |
|
Abnormal granulocytes/bands |
88 |
79 |
3 |
3 |
|
Decreased hemoglobin |
83 |
75 |
42 |
39 |
|
Abnormal lymphocytes count |
78 |
72 |
27 |
25 |
|
Pain |
45 |
41 |
44 |
39 |
|
Abnormal platelet count |
43 |
39 |
8 |
7 |
|
Abnormal alkaline phosphatase |
41 |
37 |
42 |
38 |
|
Malaise/fatigue |
37 |
34 |
16 |
14 |
|
Hyperglycemia |
33 |
31 |
32 |
30 |
|
Edema |
31 |
30 |
15 |
14 |
|
Nausea |
28 |
26 |
9 |
8 |
|
Anorexia |
24 |
22 |
16 |
14 |
|
Abnormal BUN |
24 |
22 |
22 |
20 |
|
Abnormal transaminase |
22 |
20 |
16 |
14 |
|
Alopecia |
20 |
20 |
1 |
1 |
|
Abnormal cardiac function |
19 |
18 |
0 |
0 |
|
Infection |
18 |
17 |
4 |
4 |
|
Weight loss |
18 |
17 |
13 |
12 |
|
Dyspnea |
16 |
15 |
9 |
8 |
|
Diarrhea |
16 |
14 |
4 |
4 |
|
Fever in absence of infection |
15 |
14 |
7 |
6 |
|
Weight gain |
15 |
14 |
16 |
15 |
|
Abnormal creatinine |
14 |
13 |
11 |
10 |
|
Other gastrointestinal |
13 |
14 |
11 |
11 |
|
Vomiting |
12 |
11 |
6 |
5 |
|
Other neurologic |
11 |
11 |
5 |
5 |
|
Hypocalcemia |
10 |
10 |
5 |
5 |
|
Hematuria |
9 |
11 |
5 |
6 |
|
Hyponatremia |
9 |
9 |
3 |
3 |
|
Sweats |
9 |
9 |
2 |
2 |
|
Other liver |
8 |
8 |
8 |
8 |
|
Stomatitis |
8 |
8 |
1 |
1 |
|
Cardiac dysrhythmia |
7 |
7 |
3 |
3 |
|
Hypokalemia |
7 |
7 |
4 |
4 |
|
Neuro/constipation |
7 |
7 |
2 |
2 |
|
Neuro/motor disorder |
7 |
7 |
3 |
3 |
|
Neuro/mood disorder |
6 |
6 |
2 |
2 |
|
Skin disorder |
6 |
6 |
4 |
4 |
|
Cardiac ischemia |
5 |
5 |
1 |
1 |
|
Chills |
5 |
5 |
0 |
0 |
|
Hemorrhage |
5 |
5 |
3 |
3 |
|
Myalgias/arthralgias |
5 |
5 |
3 |
3 |
|
Other kidney/bladder |
5 |
5 |
3 |
3 |
|
Other endocrine |
5 |
6 |
3 |
4 |
|
Other pulmonary |
5 |
5 |
3 |
3 |
|
Hypertension |
4 |
4 |
5 |
5 |
|
Impotence/libido |
4 |
7 |
2 |
3 |
|
Proteinuria |
4 |
6 |
2 |
3 |
|
Sterility |
3 |
5 |
2 |
3 |
General
Allergic Reaction
Hypotension, urticaria, dyspnea, and rashes have been reported occasionally. Anaphylaxis/anaphylactoid reactions have been reported rarely.
Cutaneous
Extravasation at the infusion site has been reported, which may result in erythema, swelling, pain, burning, and/or blue discoloration of the skin. Extravasation can result in tissue necrosis with resultant need for debridement and skin grafting. Phlebitis has also been reported at the site of the infusion.
Hematologic
Topoisomerase II inhibitors, including mitoxantrone, in combination with other antineoplastic agents or alone, have been associated with the development of acute leukemia (see WARNINGS).
Leukemia
Myelosuppression is rapid in onset and is consistent with the requirement to produce significant marrow hypoplasia in order to achieve a response in acute leukemia. The incidences of infection and bleeding seen in the U.S. trial are consistent with those reported for other standard induction regimens.
Hormone-Refractory Prostate Cancer
In a randomized study where dose escalation was required for neutrophil counts greater than 1000/mm3, Grade 4 neutropenia (ANC < 500/mm3) was observed in 54% of patients treated with mitoxantrone + low-dose prednisone. In a separate randomized trial where patients were treated with 14 mg/m2, Grade 4 neutropenia in 23% of patients treated with mitoxantrone + hydrocortisone was observed. Neutropenic fever/infection occurred in 11% and 10% of patients receiving mitoxantrone + corticosteroids, respectively, on the two trials. Platelets < 50,000/mm3 were noted in 4% and 3% of patients receiving mitoxantrone + corticosteroids on these trials, and there was one patient death on mitoxantrone + hydrocortisone due to intracranial hemorrhage after a fall.
Gastrointestinal
Nausea and vomiting occurred acutely in most patients and may have contributed to reports of dehydration, but were generally mild to moderate and could be controlled through the use of antiemetics. Stomatitis/mucositis occurred within 1 week of therapy.
Cardiovascular
Congestive heart failure, tachycardia, EKG changes including arrhythmias, chest pain, and asymptomatic decreases in left ventricular ejection fraction have occurred. (See WARNINGS)
Overdosage
There is no known specific antidote for mitoxantrone. Accidental overdoses have been reported. Four patients receiving 140 to 180 mg/m2 as a single bolus injection died as a result of severe leukopenia with infection. Hematologic support and antimicrobial therapy may be required during prolonged periods of severe myelosuppression.
Although patients with severe renal failure have not been studied, mitoxantrone is extensively tissue bound and it is unlikely that the therapeutic effect or toxicity would be mitigated by peritoneal or hemodialysis.
Mitoxantrone Injection Concentrate Dosage and Administration
(See also WARNINGS)
Multiple Sclerosis
The recommended dosage of mitoxantrone is 12 mg/m2 given as a short (approximately 5 to 15 minutes) intravenous infusion every 3 months. Left ventricular ejection fraction (LVEF) should be evaluated by echocardiogram or MUGA prior to administration of the initial dose of mitoxantrone and all subsequent doses. In addition, LVEF evaluations are recommended if signs or symptoms of congestive heart failure develop at any time during treatment with mitoxantrone. Mitoxantrone should not be administered to multiple sclerosis patients with an LVEF <50%, with a clinically significant reduction in LVEF, or to those who have received a cumulative lifetime dose of ≥140 mg/m2. Complete blood counts, including platelets, should be monitored prior to each course of mitoxantrone and in the event that signs or symptoms of infection develop. Mitoxantrone generally should not be administered to multiple sclerosis patients with neutrophil counts less than 1500 cells/mm3. Liver function tests should also be monitored prior to each course. Mitoxantrone therapy in multiple sclerosis patients with abnormal liver function tests is not recommended because mitoxantrone clearance is reduced by hepatic impairment and no laboratory measurement can predict drug clearance and dose adjustments.
Women with multiple sclerosis who are biologically capable of becoming pregnant, even if they are using birth control, should have a pregnancy test, and the results should be known, before receiving each dose of mitoxantrone (see WARNINGS, Pregnancy).
Hormone-Refractory Prostate Cancer
Based on data from two Phase 3 comparative trials of mitoxantrone plus corticosteroids versus corticosteroids alone, the recommended dosage of mitoxantrone is 12 to 14 mg/m2 given as a short intravenous infusion every 21 days.
Combination Initial Therapy for ANLL in Adults
For induction, the recommended dosage is 12 mg/m2 of mitoxantrone daily on Days 1 to 3 given as an intravenous infusion, and 100 mg/m2 of cytarabine for 7 days given as a continuous 24-hour infusion on Days 1 to 7.
Most complete remissions will occur following the initial course of induction therapy. In the event of an incomplete antileukemic response, a second induction course may be given. Mitoxantrone should be given for 2 days and cytarabine for 5 days using the same daily dosage levels.
If severe or life-threatening nonhematologic toxicity is observed during the first induction course, the second induction course should be withheld until toxicity resolves.
Consolidation therapy which was used in two large randomized multicenter trials consisted of mitoxantrone, 12 mg/m2 given by intravenous infusion daily on Days 1 and 2 and cytarabine, 100 mg/m2 for 5 days given as a continuous 24-hour infusion on Days 1 to 5. The first course was given approximately 6 weeks after the final induction course; the second was generally administered 4 weeks after the first. Severe myelosuppression occurred. (See CLINICAL PHARMACOLOGY)
Hepatic Impairment
For patients with hepatic impairment, there is at present no laboratory measurement that allows for dose adjustment recommendations. (See CLINICAL PHARMACOLOGY, Special Populations, Hepatic Impairment)
Preparation and Administration Precautions
MITOXANTRONE INJECTION, USP (CONCENTRATE) MUST BE DILUTED PRIOR TO USE.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. The dose of mitoxantrone should be diluted to at least 50 mL with either 0.9% Sodium Chloride Injection (USP) or 5% Dextrose Injection (USP). Mitoxantrone Injection, USP (concentrate) may be further diluted into Dextrose 5% in Water, Normal Saline or Dextrose 5% with Normal Saline and used immediately. DO NOT FREEZE.
Mitoxantrone should not be mixed in the same infusion as heparin since a precipitate may form. Because specific compatibility data are not available, it is recommended that mitoxantrone not be mixed in the same infusion with other drugs. The diluted solution should be introduced slowly into the tubing as a freely running intravenous infusion of 0.9% Sodium Chloride Injection (USP) or 5% Dextrose Injection (USP) over a period of not less than 3 minutes. Unused infusion solutions should be discarded immediately in an appropriate fashion. In the case of multiple-dose use, after penetration of the stopper, the remaining portion of the undiluted Mitoxantrone Injection, USP concentrate should be stored not longer than 7 days between 15° to 25°C (59° to 77°F) or 14 days under refrigeration. DO NOT FREEZE. CONTAINS NO PRESERVATIVE.
Care in the administration of mitoxantrone will reduce the chance of extravasation. Mitoxantrone should be administered into the tubing of a freely running intravenous infusion of 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP. The tubing should be attached to a Butterfly needle or other suitable device and inserted preferably into a large vein. If possible, avoid veins over joints or in extremities with compromised venous or lymphatic drainage. Care should be taken to avoid extravasation at the infusion site and to avoid contact of mitoxantrone with the skin, mucous membranes, or eyes. MITOXANTRONE SHOULD NOT BE ADMINISTERED SUBCUTANEOUSLY. If any signs or symptoms of extravasation have occurred, including burning, pain, pruritis, erythema, swelling, blue discoloration, or ulceration, the injection or infusion should be immediately terminated and restarted in another vein. During intravenous administration of mitoxantrone extravasation may occur with or without an accompanying stinging or burning sensation even if blood returns well on aspiration of the infusion needle. If it is known or suspected that subcutaneous extravasation has occurred, it is recommended that intermittent ice packs be placed over the area of extravasation and that the affected extremity be elevated. Because of the progressive nature of extravasation reactions, the area of injection should be frequently examined and surgery consultation obtained early if there is any sign of a local reaction.
Skin accidentally exposed to mitoxantrone should be rinsed copiously with warm water and if the eyes are involved, standard irrigation techniques should be used immediately. The use of goggles, gloves, and protective gowns is recommended during preparation and administration of the drug.
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on this subject have been published.1–4 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
References
- 1.
- NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
- 2.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm_vi_2.html.
- 3.
- American Society of Health-System Pharmacists. (2006) ASHP Guidelines on Handling Hazardous Drugs.
- 4.
- Polovich, M., White, J.M., & Kelleher, L.O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd ed.) Pittsburgh, PA: Oncology Nursing Society.
How is Mitoxantrone Injection Concentrate supplied
Mitoxantrone Injection, USP (concentrate) is a sterile aqueous solution containing mitoxantrone hydrochloride at a concentration equivalent to 2 mg mitoxantrone free base per mL supplied in vials for multiple-dose use as follows:
| Unit of Sale | Concentration |
|---|---|
|
NDC 61703-343-18
|
20 mg/10 mL |
|
NDC 61703-343-65
|
25 mg/12.5 mL |
|
NDC 61703-343-66
|
30 mg/15 mL |

Medication Guide
CONTAINS SODIUM METABISULFITE, A SULFITE THAT MAY CAUSE ALLERGIC-TYPE REACTIONS INCLUDING ANAPHYLACTIC SYMPTOMS AND LIFE-THREATENING OR LESS SEVERE ASTHMATIC EPISODES IN CERTAIN SUSCEPTIBLE PEOPLE. THE OVERALL PREVALENCE OF SULFITE SENSITIVITY IN THE GENERAL POPULATION IS UNKNOWN AND PROBABLY LOW. SULFITE SENSITIVITY IS SEEN MORE FREQUENTLY IN ASTHMATIC THAN IN NONASTHMATIC PEOPLE.
Read this Medication Guide before you start receiving MitoXANTRONE and each time you receive MitoXANTRONE. There may be new information. This information does not take the place of talking to your doctor about your medical condition or your treatment.
What is the most important information I should know about MitoXANTRONE?
MitoXANTRONE can cause serious side effects, including:
- •
-
decrease in the ability of your bone marrow to make blood cells (myelosuppression). Your doctor may do blood tests during treatment with MitoXANTRONE to check your blood cell counts. The symptoms of myelosuppression can include:
- o
- feeling tired
- o
- increased infections
- o
- bruising and bleeding easily
- •
-
heart problems (congestive heart failure) that may lead to death even in people who have never had heart problems before. Heart failure can happen while you receive MitoXANTRONE, or months to years after you stop receiving MitoXANTRONE. Your risk of heart failure increases the more MitoXANTRONE you receive.
Call your doctor or get medical help right away if you have any of these problems during or after treatment with MitoXANTRONE: Before receiving MitoXANTRONE for the first time, you should have the following tests done:If you receive MitoXANTRONE to treat Multiple Sclerosis (MS), your doctor should also do the tests above:- o
- shortness of breath
- o
- swelling of your ankles or feet
- o
- sudden weight gain
- o
- fast heartbeat or pounding in your chest
- o
- physical examination
- o
- a test to check your heart's electrical activity (electrocardiogram)
- o
- a test to check your heart's ability to pump blood
- o
- before you receive each MitoXANTRONE dose
- o
- yearly after you stop receiving MitoXANTRONE treatment
- •
- acute myeloid leukemia (AML). Receiving MitoXANTRONE increases your risk of AML. AML is a cancer of the blood-forming cells of your bone marrow. Symptoms of AML can include:
|
|
- •
- skin problems at your injection site. If MitoXANTRONE leaks out of your vein, skin problems can happen that may lead to serious skin damage (necrosis). Necrosis may need to be repaired surgically. Tell your doctor right away if you have any of the following problems at your injection site:
|
|
What is MitoXANTRONE?
MitoXANTRONE is a prescription medicine used alone or with other medicines to treat people with:
- •
- secondary (chronic) progressive, progressive relapsing, or worsening relapsing-remitting multiple sclerosis (MS)
- •
- pain related to advanced hormone-refractory prostate cancer
- •
- acute nonlymphocytic leukemia (ANLL)
MitoXANTRONE is not for people with primary progressive MS. It is not known if MitoXANTRONE is safe and effective in children.
Who should not receive MitoXANTRONE?
Do not receive MitoXANTRONE if you are allergic to MitoXANTRONE or any of the ingredients in MitoXANTRONE. See the end of this Medication Guide for a complete list of ingredients in MitoXANTRONE.
What should I tell my doctor before receiving MitoXANTRONE?
Before you receive MitoXANTRONE, tell your doctor if you have:
- •
- received MitoXANTRONE in the past
- •
- heart problems
- •
- liver problems
- •
- kidney problems
- •
- low blood cell counts
- •
- an infection
- •
- had radiation treatment in your chest area
- •
- any other medical conditions
- •
- are pregnant or plan to become pregnant. MitoXANTRONE may harm your unborn baby. Women who are able to become pregnant should use effective birth control (contraception) while using MitoXANTRONE and should have a pregnancy test, with known results, before receiving each dose of MitoXANTRONE. Talk to your doctor about using effective birth control while you receive MitoXANTRONE.
- •
- are breastfeeding or plan to breastfeed. MitoXANTRONE can pass into your breast milk and may harm your baby. Talk to your doctor about the best way to feed your baby if you receive MitoXANTRONE. Do not breastfeed while receiving MitoXANTRONE.
Tell your doctor about all the medicines you take, including prescription and nonprescription medicines, vitamins, and herbal supplements.
Using MitoXANTRONE with certain other medicines may cause serious side effects.
Especially tell your doctor if you take or have taken:
- •
- medicines for cancer treatment called anthracyclines or anthracenediones
- •
- medicines that may affect your heart
Ask your doctor or pharmacist for a list of these medicines if you are not sure if you take or have taken any of these medicines.
Know the medicines you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine.
How should I receive MitoXANTRONE?
- •
- MitoXANTRONE is given by slow infusion through a needle placed in a vein (intravenous infusion) in your arm.
- •
- Your doctor will tell you how often you will receive MitoXANTRONE.
- •
- If you receive MitoXANTRONE to treat MS, your doctor should check how well your heart is working before each MitoXANTRONE dose. Talk to your doctor if you have not had your heart tests done before your MitoXANTRONE dose.
- •
- Your doctor will do blood tests during your treatment with MitoXANTRONE to check your blood cell counts.
- •
- If you are a woman of childbearing age taking MitoXANTRONE to treat MS, your doctor should do a pregnancy test before each MitoXANTRONE dose, even if you are using birth control.
- •
- If you receive MitoXANTRONE to treat MS, there is a limit to the total amount of MitoXANTRONE you can receive during your lifetime. There is a higher risk of heart failure with increasing total lifetime doses of MitoXANTRONE.
What are the possible side effects of MitoXANTRONE?
MitoXANTRONE may cause serious side effects, including:
- •
- See "What is the most important information I should know about MitoXANTRONE?" The most common side effects of MitoXANTRONE include:
|
||
|
|
|
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of MitoXANTRONE. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of MitoXANTRONE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide.
This Medication Guide summarizes the most important information about MitoXANTRONE. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about MitoXANTRONE that is written for health professionals.
For more information go to www.hospira.com or call 1-800-615-0187.
What are the ingredients in MitoXANTRONE?
Active ingredient: MitoXANTRONE hydrochloride
Inactive ingredients: sodium chloride, sodium metabisulfite, sodium acetate, and acetic acid
This Medication Guide has been approved by the U.S. Food and Drug Administration.

Distributed by Hospira Inc., Lake Forest, IL 60045 USA
LAB-1312-1.0
Revised: 5/2018
PRINCIPAL DISPLAY PANEL - 10 mL Vial Label
10 mL Vial
NDC 61703-343-18
Sterile
MitoXANTRONE Injection, USP
(concentrate)
20 mg/ 10 mL
(2 mg/mL)
Rx only
For IV Infusion After Dilution
Multiple-dose Vial Cytotoxic Agent
Pharmacist dispense enclosed medication guide to each patient.
Hospira
PRINCIPAL DISPLAY PANEL - 10 mL Vial Carton
VIAL
Hospira
1 x 10 mL Vial
NDC 61703-343-18
Sterile
Rx only
MitoXANTRONE
Injection, USP
(concentrate)
20 mg/ 10 mL
(2 mg/mL)
For Intravenous Infusion
After Dilution
Multiple-dose Vial
Pharmacist dispense enclosed
medication guide to each
patient.
Cytotoxic Agent
PRINCIPAL DISPLAY PANEL - 12.5 mL Vial Label
12.5 mL Vial
NDC 61703-343-65
Sterile
MitoXANTRONE
Injection, USP
(concentrate)
25 mg/ 12.5 mL
(2 mg/mL)
Rx only
For IV Infusion After Dilution
Multiple-dose Vial
Cytotoxic Agent
Pharmacist dispense enclosed
medication guide to each patient.
PRINCIPAL DISPLAY PANEL - 12.5 mL Vial Carton
VIAL
Hospira
1 x 12.5 mL Vial
NDC 61703-343-65
Sterile
Rx only
MitoXANTRONE
Injection, USP
(concentrate)
25 mg/ 12.5 mL
(2 mg/mL)
For Intravenous Infusion
After Dilution
Multiple-dose Vial
Pharmacist dispense
enclosed medication guide to
each patient.
Cytotoxic Agent



| MITOXANTRONE
mitoxantrone injection, solution, concentrate |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - Hospira, Inc. (141588017) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Zydus Hospira Oncology Private Limited | 676190889 | ANALYSIS(61703-343) , MANUFACTURE(61703-343) , PACK(61703-343) , LABEL(61703-343) | |
More about mitoxantrone
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (2)
- Side effects
- Dosage information
- During pregnancy
- Drug class: antibiotics/antineoplastics
- Breastfeeding
- En español