Kisunla: Package Insert / Prescribing Info
Package insert / product label
Generic name: donanemab-azbt
Dosage form: injection, solution
Drug class: Miscellaneous central nervous system agents
J Code (medical billing code): J0175 (2 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jul 17, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
KISUNLA (donanemab-azbt) injection, for intravenous use
Initial U.S. Approval: 2024
WARNING: AMYLOID RELATED IMAGING ABNORMALITIES
See full prescribing information for complete boxed warning.
Monoclonal antibodies directed against aggregated forms of beta amyloid, including KISUNLA, can cause amyloid related imaging abnormalities (ARIA), as ARIA with edema (ARIA-E) and ARIA with hemosiderin deposition (ARIA-H). ARIA is usually asymptomatic, although serious and life-threatening events can rarely occur. Serious intracerebral hemorrhages >1 cm have occurred in patients treated with this class of medications. ARIA-E can cause focal neurologic deficits that can mimic ischemic stroke. (5.1, 6.1)
ApoE ε4 Homozygotes
Patients treated with this class of medications, including KISUNLA, who are ApoE ε4 homozygotes have a higher incidence of ARIA, including symptomatic and serious ARIA, compared to heterozygotes and noncarriers. Testing for ApoE ε4 status should be performed prior to initiation of treatment to inform the risk of developing ARIA. Prior to testing, prescribers should discuss with patients the risk of ARIA across genotypes and the implications of genetic testing results. (5.1)
Consider the benefit for the treatment of Alzheimer's disease and risk of ARIA when deciding to treat with KISUNLA. (5.1, 14)
Indications and Usage for Kisunla
KISUNLA is an amyloid beta-directed antibody indicated for the treatment of Alzheimer's disease. Treatment with KISUNLA should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in the clinical trials. (1)
Kisunla Dosage and Administration
- Confirm the presence of amyloid beta pathology prior to initiating treatment. (2.1)
- The recommended dosage of KISUNLA is 700 mg administered as an intravenous infusion over approximately 30 minutes every four weeks for the first three doses, followed by 1400 mg every four weeks. (2.2)
- Consider stopping dosing with KISUNLA based on reduction of amyloid plaques to minimal levels on amyloid PET imaging. (2.2)
- Obtain a recent baseline brain MRI prior to initiating treatment. (2.3, 5.1)
- Obtain an MRI prior to the 2nd, 3rd, 4th, and 7th infusions. If radiographically observed ARIA occurs, treatment recommendations are based on type, severity, and presence of symptoms. (2.3, 5.1)
- Dilution to a final concentration of 4 mg/mL to 10 mg/mL with 0.9% Sodium Chloride Injection, is required prior to administration. (2.4)
Dosage Forms and Strengths
Injection: 350 mg/20 mL (17.5 mg/mL) in a single-dose vial. (3)
Contraindications
Warnings and Precautions
- Amyloid Related Imaging Abnormalities (ARIA): Enhanced clinical vigilance for ARIA is recommended during the first 24 weeks of treatment with KISUNLA. Risk of ARIA, including symptomatic ARIA, was increased in apolipoprotein E ε4 (ApoE ε4) homozygotes compared to heterozygotes and noncarriers. The risk of ARIA-E and ARIA-H is increased in KISUNLA-treated patients with pretreatment microhemorrhages and/or superficial siderosis. If a patient experiences symptoms suggestive of ARIA, clinical evaluation should be performed, including MRI scanning if indicated. (2.3, 5.1)
- Infusion-Related Reactions: The infusion rate may be reduced, or the infusion may be discontinued, and appropriate therapy initiated as clinically indicated. Consider pre-treatment with antihistamines, acetaminophen, or corticosteroids prior to subsequent dosing. (5.3)
Adverse Reactions/Side Effects
Most common adverse reactions (at least 10% and higher incidence compared to placebo): ARIA-E, ARIA-H microhemorrhage, ARIA-H superficial siderosis, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eli Lilly and Company at 1-800-LillyRx (1-800-545-5979) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2024
Full Prescribing Information
WARNING: AMYLOID RELATED IMAGING ABNORMALITIES
Monoclonal antibodies directed against aggregated forms of beta amyloid, including KISUNLA, can cause amyloid related imaging abnormalities (ARIA), characterized as ARIA with edema (ARIA-E) and ARIA with hemosiderin deposition (ARIA-H). Incidence and timing of ARIA vary among treatments. ARIA usually occurs early in treatment and is usually asymptomatic, although serious and life-threatening events rarely can occur. Serious intracerebral hemorrhages >1 cm, some of which have been fatal, have been observed in patients treated with this class of medications. Because ARIA-E can cause focal neurologic deficits that can mimic an ischemic stroke, treating clinicians should consider whether such symptoms could be due to ARIA-E before giving thrombolytic therapy in a patient being treated with KISUNLA [see Warnings and Precautions (5.1), Adverse Reactions (6.1)].
ApoE ε4 Homozygotes
Patients who are apolipoprotein E ε4 (ApoE ε4) homozygotes (approximately 15% of Alzheimer's disease patients) treated with this class of medications, including KISUNLA, have a higher incidence of ARIA, including symptomatic, serious, and severe radiographic ARIA, compared to heterozygotes and noncarriers [see Warnings and Precautions (5.1)]. Testing for ApoE ε4 status should be performed prior to initiation of treatment to inform the risk of developing ARIA. Prior to testing, prescribers should discuss with patients the risk of ARIA across genotypes and the implications of genetic testing results. Prescribers should inform patients that if genotype testing is not performed, they can still be treated with KISUNLA; however, it cannot be determined if they are ApoE ε4 homozygotes and at higher risk for ARIA [see Warnings and Precautions (5.1)].
Consider the benefit of KISUNLA for the treatment of Alzheimer's disease and potential risk of serious adverse events associated with ARIA when deciding to initiate treatment with KISUNLA [see Warnings and Precautions (5.1) and Clinical Studies (14)].
1. Indications and Usage for Kisunla
KISUNLATM is indicated for the treatment of Alzheimer's disease. Treatment with KISUNLA should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in the clinical trials.
2. Kisunla Dosage and Administration
2.1 Patient Selection
Confirm the presence of amyloid beta pathology prior to initiating treatment [see Clinical Pharmacology (12.1)].
2.2 Dosing Instructions
The recommended dosage of KISUNLA is 700 mg every four weeks for three doses, then 1400 mg every four weeks (see Table 1). KISUNLA is administered every four weeks as an intravenous infusion over approximately 30 minutes. KISUNLA must be diluted prior to administration (see Table 4).
| Intravenous Infusion
(every 4 weeks) | KISUNLA Dosage
(administered over approximately 30 minutes) |
| Infusions 1, 2, and 3 | 700 mg |
| Infusion 4 and beyond | 1400 mg |
Consider stopping dosing with KISUNLA based on reduction of amyloid plaques to minimal levels on amyloid PET imaging. In Study 1, dosing was stopped based on a reduction of amyloid levels below predefined thresholds on PET imaging [see Clinical Studies (14)].
If an infusion is missed, resume administration every 4 weeks at the same dose as soon as possible.
2.3 Monitoring and Dosing Interruption for Amyloid Related Imaging Abnormalities
KISUNLA can cause amyloid related imaging abnormalities -edema (ARIA-E) and -hemosiderin deposition (ARIA-H) [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Monitoring for ARIA
Obtain a recent baseline brain magnetic resonance imaging (MRI) prior to initiating treatment with KISUNLA. Obtain an MRI prior to the 2nd, 3rd, 4th, and 7th infusions. If a patient experiences symptoms suggestive of ARIA, clinical evaluation should be performed, including an MRI if indicated.
Recommendations for Dosing Interruptions in Patients with ARIA
ARIA-E
The recommendations for dosing interruptions for patients with ARIA-E are provided in Table 2.
|
a Mild: discomfort noticed, but no disruption of normal daily activity. |
|||
|
b Suspend until MRI demonstrates radiographic resolution and symptoms, if present, resolve; consider a follow-up MRI to assess for resolution 2 to 4 months after initial identification. Resumption of dosing should be guided by clinical judgment. |
|||
| Clinical Symptom Severitya | ARIA-E Severity on MRI | ||
| Mild | Moderate | Severe | |
| Asymptomatic | May continue dosing at current dose and schedule | Suspend dosingb | Suspend dosingb |
| Mild | May continue dosing based on clinical judgment | Suspend dosingb | |
| Moderate or Severe | Suspend dosingb | ||
ARIA-H
The recommendations for dosing interruptions for patients with ARIA-H are provided in Table 3.
|
a Suspend until MRI demonstrates radiographic stabilization and symptoms, if present, resolve; resumption of dosing should be guided by clinical judgment; consider a follow-up MRI to assess for stabilization 2 to 4 months after initial identification. |
|||
|
b Suspend until MRI demonstrates radiographic stabilization and symptoms, if present, resolve. Use clinical judgment when considering whether to continue treatment or permanently discontinue KISUNLA. |
|||
| Clinical Symptom Severity | ARIA-H Severity on MRI | ||
| Mild | Moderate | Severe | |
| Asymptomatic | May continue dosing at current dose and schedule | Suspend dosinga | Suspend dosingb |
| Symptomatic | Suspend dosinga | Suspend dosinga | |
In patients who develop intracerebral hemorrhage greater than 1 cm in diameter during treatment with KISUNLA, suspend dosing until MRI demonstrates radiographic stabilization and symptoms, if present, resolve. Resumption of dosing should be guided by clinical judgment.
2.4 Dilution Instructions
- Prior to administration, KISUNLA must be diluted with 0.9% sodium chloride injection (see Table 4).
- Use aseptic technique when preparing the diluted KISUNLA solution for intravenous infusion.
- Allow KISUNLA to equilibrate to room temperature before preparation.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. KISUNLA solution is clear to opalescent, colorless to slightly yellow to slightly brown. Do not use if particulate matter or discolorations are present.
- Withdraw required volume of KISUNLA and mix with 0.9% sodium chloride injection, to the recommended total volume for a final concentration of 4 mg/mL to 10 mg/mL (see Table 4). Use only 0.9% sodium chloride injection for dilution.
|
a final concentration of 4 mg/mL to 10 mg/mL |
||||
|
b 2 vials of KISUNLA |
||||
|
c 4 vials of KISUNLA |
||||
| KISUNLA Dose (mg) | KISUNLA Volume (mL) | Volume of 0.9% Sodium Chloride Injection Diluent (mL) | Final Volume of Diluted Solution to be Infused (mL) | Final Concentration of Diluted Solution (mg/mL)a |
| 700 mg | 40 mLb | 30 mL to 135 mL | 70 mL to 175 mL | 700 mg/175 mL (4 mg/mL) to 700 mg/70 mL (10 mg/mL) |
| 1400 mg | 80 mLc | 60 mL to 270 mL | 140 mL to 350 mL | 1400 mg/350 mL (4 mg/mL) to 1400 mg/140 mL (10 mg/mL) |
- Each vial is for one-time use only. Discard any unused portion left in the vial.
- Gently invert the diluted KISUNLA solution to mix completely. Do not shake.
- After dilution, immediate use is recommended [see Description (11)]. If the diluted KISUNLA solution is not administered immediately, store refrigerated at 2°C to 8°C (36°F to 46°F) for up to 72 hours or at room temperature (20°C to 25°C [68°F to 77°F]) for up to 12 hours.
- Do not freeze the diluted KISUNLA solution.
- Storage times include the duration of infusion.
2.5 Administration Instructions
- Visually inspect the diluted KISUNLA solution for particles or discoloration prior to administration. Do not use if it is discolored, or opaque or foreign particles are seen.
- Prior to infusion, if the diluted solution has been stored under refrigeration, allow the diluted KISUNLA solution to warm to room temperature.
- Administer the entire diluted solution intravenously over approximately 30 minutes.
- Promptly discontinue the infusion upon the first observation of any signs or symptoms consistent with a hypersensitivity-type reaction [see Warnings and Precautions (5.2)].
- Flush the line only with 0.9% sodium chloride injection at the end of the infusion per access specific line maintenance protocol.
- Observe the patient post-infusion for a minimum of 30 minutes to evaluate for infusion reactions and hypersensitivity reactions [see Warnings and Precautions (5.2)].
3. Dosage Forms and Strengths
Injection: 350 mg/20 mL (17.5 mg/mL) clear to opalescent, colorless to slightly yellow to slightly brown solution in a single-dose vial.
4. Contraindications
KISUNLA is contraindicated in patients with known serious hypersensitivity to donanemab-azbt or to any of the excipients. Reactions have included anaphylaxis [see Warnings and Precautions (5.2)].
5. Warnings and Precautions
5.1 Amyloid Related Imaging Abnormalities
Monoclonal antibodies directed against aggregated forms of beta amyloid, including KISUNLA, can cause amyloid related imaging abnormalities (ARIA), characterized as ARIA with edema (ARIA-E), which can be observed on MRI as brain edema or sulcal effusions, and ARIA with hemosiderin deposition (ARIA-H), which includes microhemorrhage and superficial siderosis. ARIA can occur spontaneously in patients with Alzheimer’s disease, particularly in patients with MRI findings suggestive of cerebral amyloid angiopathy, such as pretreatment microhemorrhage or superficial siderosis. ARIA-H associated with monoclonal antibodies directed against aggregated forms of beta amyloid generally occurs in association with an occurrence of ARIA-E. ARIA-H of any cause and ARIA-E can occur together.
ARIA usually occurs early in treatment and is usually asymptomatic, although serious and life-threatening events, including seizure and status epilepticus, rarely can occur. When present, reported symptoms associated with ARIA may include, but are not limited to, headache, confusion, visual changes, dizziness, nausea, and gait difficulty. Focal neurologic deficits may also occur. Symptoms associated with ARIA usually resolve over time. In addition to ARIA, intracerebral hemorrhages greater than 1 cm in diameter have occurred in patients treated with KISUNLA.
Consider the benefit of KISUNLA for the treatment of Alzheimer's disease and potential risk of serious adverse events associated with ARIA when deciding to initiate treatment with KISUNLA.
Incidence of ARIA
Symptomatic ARIA occurred in 6% (52/853) of patients treated with KISUNLA in Study 1 [see Adverse Reactions (6.1)]. Clinical symptoms associated with ARIA resolved in approximately 85% (44/52) of patients.
Including asymptomatic radiographic events, ARIA was observed in 36% (307/853) of patients treated with KISUNLA, compared to 14% (122/874) of patients on placebo in Study 1. ARIA-E was observed in 24% (201/853) of patients treated with KISUNLA compared with 2% (17/874) of patients on placebo. ARIA-H was observed in 31% (263/853) of patients treated with KISUNLA compared with 13% (111/874) of patients on placebo. There was no increase in isolated ARIA-H (i.e., ARIA-H in patients who did not also experience ARIA-E) for KISUNLA compared to placebo.
Incidence of Intracerebral Hemorrhage
Intracerebral hemorrhage greater than 1 cm in diameter was reported in 0.5% (4/853) of patients in Study 1 after treatment with KISUNLA compared to 0.2% (2/874) of patients on placebo. Fatal events of intracerebral hemorrhage in patients taking KISUNLA have been observed.
Risk Factors for ARIA and Intracerebral Hemorrhage
ApoE ε4 Carrier Status
The risk of ARIA, including symptomatic and serious ARIA, is increased in apolipoprotein E ε4 (ApoE ε4) homozygotes. Approximately 15% of Alzheimer’s disease patients are ApoE ε4 homozygotes. In Study 1, 17% (143/850) of patients in the KISUNLA arm were apolipoprotein E ε4 (ApoE ε4) homozygotes, 53% (452/850) were heterozygotes, and 30% (255/850) were noncarriers. The incidence of ARIA was higher in ApoE ε4 homozygotes (55% on KISUNLA vs. 22% on placebo) than in heterozygotes (36% on KISUNLA vs. 13% on placebo) and noncarriers (25% on KISUNLA vs. 12% on placebo). Among patients treated with KISUNLA, symptomatic ARIA-E occurred in 8% of ApoE ε4 homozygotes compared with 7% of heterozygotes and 4% of noncarriers. Serious events of ARIA occurred in 3% of ApoE ε4 homozygotes, 2% of heterozygotes and 1% of noncarriers. The recommendations for management of ARIA do not differ between ApoE ε4 carriers and noncarriers [see Dosage and Administration (2.3)]. Testing for ApoE ε4 status should be performed prior to initiation of treatment to inform the risk of developing ARIA. Prior to testing, prescribers should discuss with patients the risk of ARIA across genotypes and the implications of genetic testing results. Prescribers should inform patients that if genotype testing is not performed, they can still be treated with KISUNLA; however, it cannot be determined if they are ApoE ε4 homozygotes and at a higher risk for ARIA. An FDA-authorized test for detection of ApoE ε4 alleles to identify patients at risk of ARIA if treated with KISUNLA is not currently available. Currently available tests used to identify ApoE ε4 alleles may vary in accuracy and design.
Radiographic Findings of Cerebral Amyloid Angiopathy (CAA)
Neuroimaging findings that may indicate CAA include evidence of prior intracerebral hemorrhage, cerebral microhemorrhage, and cortical superficial siderosis. CAA has an increased risk for intracerebral hemorrhage. The presence of an ApoE ε4 allele is also associated with cerebral amyloid angiopathy.
In Study 1, the baseline presence of at least 2 microhemorrhages or the presence of at least 1 area of superficial siderosis on MRI, which may be suggestive of CAA, were identified as risk factors for ARIA. Patients were excluded from enrollment in Study 1 for findings on neuroimaging of prior intracerebral hemorrhage greater than 1 cm in diameter, more than 4 microhemorrhages, more than 1 area of superficial siderosis, severe white matter disease, and vasogenic edema.
Concomitant Antithrombotic or Thrombolytic Medication
In Study 1, baseline use of antithrombotic medication (aspirin, other antiplatelets, or anticoagulants) was allowed. The majority of exposures to antithrombotic medications were to aspirin. The incidence of ARIA-H was 30% (106/349) in patients taking KISUNLA with a concomitant antithrombotic medication within 30 days compared to 29% (148/504) who did not receive an antithrombotic within 30 days of an ARIA-H event. The incidence of intracerebral hemorrhage greater than 1 cm in diameter was 0.6% (2/349 patients) in patients taking KISUNLA with a concomitant antithrombotic medication compared to 0.4% (2/504) in those who did not receive an antithrombotic. The number of events and the limited exposure to non-aspirin antithrombotic medications limit definitive conclusions about the risk of ARIA or intracerebral hemorrhage in patients taking antithrombotic medications.
One fatal intracerebral hemorrhage occurred in a patient taking KISUNLA in the setting of focal neurologic symptoms of ARIA and the use of a thrombolytic agent. Additional caution should be exercised when considering the administration of antithrombotics or a thrombolytic agent (e.g., tissue plasminogen activator) to a patient already being treated with KISUNLA. Because ARIA-E can cause focal neurologic deficits that can mimic an ischemic stroke, treating clinicians should consider whether such symptoms could be due to ARIA-E before giving thrombolytic therapy in a patient being treated with KISUNLA.
Caution should be exercised when considering the use of KISUNLA in patients with factors that indicate an increased risk for intracerebral hemorrhage and in particular for patients who need to be on anticoagulant therapy or patients with findings on MRI that are suggestive of cerebral amyloid angiopathy.
Radiographic Severity
The radiographic severity of ARIA associated with KISUNLA was classified by the criteria shown in Table 5.
|
a Includes new or worsening superficial siderosis. |
|||
| ARIA Type | Radiographic Severity | ||
| Mild | Moderate | Severe | |
| ARIA-E | FLAIR hyperintensity confined to sulcus and/or cortex/subcortex white matter in one location <5 cm. | FLAIR hyperintensity 5 to 10 cm in single greatest dimension, or more than 1 site of involvement, each measuring <10 cm. | FLAIR hyperintensity >10 cm with associated gyral swelling and sulcal effacement. One or more separate/independent sites of involvement may be noted. |
| ARIA-H microhemorrhage | Less than or equal to 4 new incident microhemorrhages | 5 to 9 new incident microhemorrhages | 10 or more new incident microhemorrhages |
| ARIA-H superficial siderosis | 1 newa focal area of superficial siderosis | 2 new focal areas of superficial siderosis | Greater than 2 new focal areas of superficial siderosis |
The majority of ARIA-E radiographic events in Study 1 occurred early in treatment (within the first 24 weeks), although ARIA can occur at any time and patients can have more than one episode. The maximum radiographic severity of ARIA-E in patients treated with KISUNLA was mild in 7% (59/853) of patients, moderate in 15% (128/853) of patients, and severe in 2% (14/853) of patients. Resolution on MRI after the first ARIA-E event occurred in 63% of patients treated with KISUNLA by 12 weeks, 80% by 20 weeks, and 83% overall after detection. The maximum radiographic severity of ARIA-H microhemorrhage in patients treated with KISUNLA was mild in 17% (143/853) of patients, moderate in 4% (34/853) of patients, and severe in 5% (40/853) of patients. The maximum radiographic severity of ARIA-H superficial siderosis in patients treated with KISUNLA was mild in 6% (47/853) of patients, moderate in 4% (32/853) of patients, and severe in 5% (46/853) of patients. Among patients treated with KISUNLA, the rate of severe radiographic ARIA-E was highest in ApoE ε4 homozygotes 3% (4/143) compared to heterozygotes 2% (9/452) or noncarriers 0.4% (1/255). Among patients treated with KISUNLA, the rate of severe radiographic ARIA-H was highest in ApoE ε4 homozygotes 22% (31/143) compared to heterozygotes 8% (38/452) or noncarriers 4% (9/255).
Monitoring and Dose Management Guidelines
Recommendations for dosing in patients with ARIA-E depend on clinical symptoms and radiographic severity [see Dosage and Administration (2.3)]. Recommendations for dosing in patients with ARIA-H depend on the type of ARIA-H and radiographic severity [see Dosage and Administration (2.3)]. Use clinical judgment in considering whether to continue dosing in patients with recurrent ARIA-E.
Baseline brain MRI and periodic monitoring with MRI are recommended [see Dosage and Administration (2.3)]. Enhanced clinical vigilance for ARIA is recommended during the first 24 weeks of treatment with KISUNLA. If a patient experiences symptoms suggestive of ARIA, clinical evaluation should be performed, including MRI if indicated. If ARIA is observed on MRI, careful clinical evaluation should be performed prior to continuing treatment.
There is limited experience in patients who continued dosing through asymptomatic but radiographically mild to moderate ARIA-E. There are limited data for dosing patients who have experienced recurrent episodes of ARIA-E.
Providers should encourage patients to participate in real world data collection (e.g., registries) to help further the understanding of Alzheimer's disease and the impact of Alzheimer's disease treatments. Providers and patients can contact 1-800-LillyRx (1-800-545-5979) for a list of currently enrolling programs.
5.2 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis and angioedema, have occurred in patients who were treated with KISUNLA. Promptly discontinue the infusion upon the first observation of any signs or symptoms consistent with a hypersensitivity reaction and initiate appropriate therapy. KISUNLA is contraindicated in patients with a history of serious hypersensitivity to donanemab-azbt or to any of the excipients of KISUNLA.
5.3 Infusion-Related Reactions
In Study 1, infusion-related reactions were observed in 9% (74/853) of patients treated with KISUNLA compared to 0.5% (4/874) of patients on placebo; the majority (70%, 52/74) occurred within the first 4 infusions. Infusion reactions typically occur during infusion or within 30 minutes post-infusion. Infusion-related reactions were mostly mild (57%) or moderate (39%) in severity. Infusion-related reactions resulted in discontinuations in 4% (31/853) of patients treated with KISUNLA. Signs and symptoms of infusion-related reactions include chills, erythema, nausea/vomiting, difficulty breathing/dyspnea, sweating, elevated blood pressure, headache, chest pain, and low blood pressure.
In the event of an infusion-related reaction, the infusion rate may be reduced, or the infusion may be discontinued, and appropriate therapy initiated as clinically indicated. Pre-treatment with antihistamines, acetaminophen, or corticosteroids prior to subsequent dosing may be considered.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Amyloid Related Imaging Abnormalities [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- Infusion-Related Reactions [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of KISUNLA has been evaluated in 2885 patients with Alzheimer's disease who received at least one dose of KISUNLA intravenously. In the clinical studies of KISUNLA, 1912 patients with Alzheimer's disease received KISUNLA once monthly for at least 6 months, 1057 patients for at least 12 months, and 432 patients for at least 18 months, at the recommended dosing schedule.
In Study 1 (NCT04437511), a total of 853 patients with Alzheimer's disease received at least one dose of KISUNLA [see Clinical Studies (14)].
Thirteen percent of patients treated with KISUNLA compared to 4% of patients on placebo stopped study treatment because of an adverse reaction. The most common adverse reaction leading to discontinuation of KISUNLA was infusion-related reaction (4% of patients treated with KISUNLA compared to no patient on placebo).
Table 6 shows adverse reactions that were reported in at least 5% of patients treated with KISUNLA and at least 2% more frequently than in patients on placebo in Study 1.
|
a As assessed by MRI. A participant could have both microhemorrhage and superficial siderosis. |
||
| Adverse Reaction | KISUNLA
N = 853 % | Placebo
N = 874 % |
| ARIA-H microhemorrhagea | 25 | 11 |
| ARIA-E | 24 | 2 |
| ARIA-H superficial siderosisa | 15 | 3 |
| Headache | 13 | 10 |
| Infusion-related reaction | 9 | 0.5 |
Less Common Adverse Reactions
Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis, occurred in 3% of patients treated with KISUNLA compared to 0.7% of patients on placebo [see Warnings and Precautions (5.2)].
Intestinal Obstruction and Intestinal Perforation
Three patients (0.4%) treated with KISUNLA had serious adverse reactions of intestinal obstruction compared to no patients on placebo. Two patients (0.2%) treated with KISUNLA had serious adverse reactions of intestinal perforation compared to one patient (0.1%) on placebo.
Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions
In Study 1, approximately 10% of KISUNLA-treated patients who developed anti-drug antibodies (ADA) reported infusion-related reactions compared to 2% of patients who did not develop ADA [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.6)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate data on KISUNLA use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. No animal studies have been conducted to assess the potential reproductive or developmental toxicity of KISUNLA.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
8.2 Lactation
Risk Summary
There are no data on the presence of donanemab-azbt in human milk, the effects on the breastfed infant, or the effects of the drug on milk production. Published data from other monoclonal antibodies generally indicate low passage of monoclonal antibodies into human milk and limited systemic exposure in the breastfed infant. The effects of this limited exposure are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for KISUNLA and any potential adverse effects on the breastfed infant from KISUNLA or from the underlying maternal condition.
8.5 Geriatric Use
In Study 1, the age of patients exposed to KISUNLA ranged from 59 to 86 years, with a mean age of 73 years; 90% were 65 years and older, and 41% were 75 years and older. No overall differences in safety or effectiveness of KISUNLA have been observed between patients 65 years of age and older and younger adult patients.
11. Kisunla Description
Donanemab-azbt is a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against insoluble N-truncated pyroglutamate amyloid beta, and is expressed in a Chinese hamster ovary cell line. Donanemab-azbt has an approximate molecular weight of 145 kDa.
KISUNLA (donanemab-azbt) injection is a sterile, preservative-free, clear to opalescent, colorless to slightly yellow to slightly brown solution for intravenous infusion after dilution. KISUNLA is supplied in single-dose vials available in a concentration of 350 mg/20 mL (17.5 mg/mL).
Each mL of solution contains 17.5 mg donanemab-azbt, anhydrous citric acid (0.32 mg), polysorbate 80 (0.20 mg), sodium citrate (2.15 mg), sucrose (80 mg), and Water for Injection, USP, at a pH of 5.5 to 6.5.
12. Kisunla - Clinical Pharmacology
12.1 Mechanism of Action
Donanemab-azbt is a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against insoluble N-truncated pyroglutamate amyloid beta. The accumulation of amyloid beta plaques in the brain is a defining pathophysiological feature of Alzheimer's disease. Donanemab-azbt reduces amyloid beta plaques, as evaluated in Study 1 [see Clinical Studies (14)].
12.2 Pharmacodynamics
Effect of KISUNLA on Amyloid Beta Pathology
The effect of KISUNLA on amyloid beta plaque levels in the brain was evaluated using amyloid Positron Emission Tomography (PET) imaging (18F-florbetapir tracer). The PET signal was quantified using the Standard Uptake Value Ratio (SUVR) method to estimate brain levels of amyloid beta plaque in composites of brain areas expected to be widely affected by Alzheimer's disease pathology (precuneus, frontal, anterior cingulate, posterior cingulate, parietal, and temporal cortices), compared to a brain region expected to be spared of such pathology (cerebellum). Results of amyloid PET were also expressed on the Centiloid scale.
In Study 1 [see Clinical Studies (14)], KISUNLA reduced amyloid beta plaque levels in the brain in a time-dependent manner, starting at Week 24, and continuing through Week 76 (p<0.0001), compared to placebo (see Figure 1 and Table 7). In clinical pharmacology studies, KISUNLA demonstrated a dose- and time-dependent reduction in amyloid beta plaque, with the decrease observed starting at Week 12.
Figure 1: Reduction in Brain Amyloid Beta Plaque (Change from Baseline) on Amyloid Beta PET Imaging Composite (SUVR and Centiloids) in Study 1a
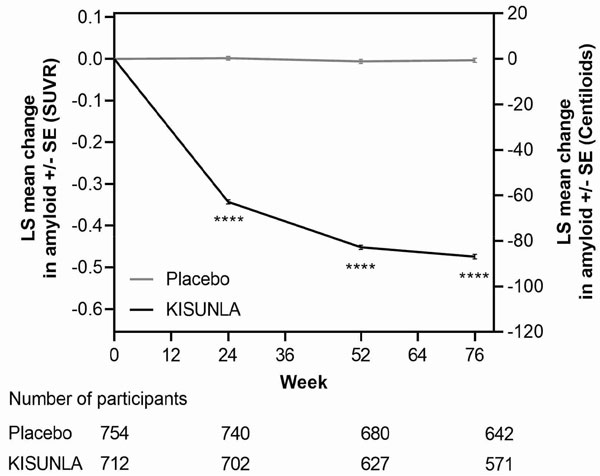
a ****p<0.0001.
During an off-treatment period, amyloid PET values began to increase with a median rate of 2.80 Centiloids/year.
Effect of KISUNLA on Tau Pathophysiology
A reduction in plasma p-tau217 was observed with KISUNLA compared to placebo in Study 1 (see Table 7).
|
N is the number of patients with baseline value. |
||
|
a Results should be interpreted with caution due to the uncertainties in bioanalysis. |
||
| Biomarker Endpoint at Week 76 | KISUNLA | Placebo |
| Amyloid Beta PET SUVR | N = 712 | N = 754 |
| Mean baseline | 1.53 | 1.52 |
| Adjusted mean change from baseline | -0.47 | -0.00 |
| Difference from placebo | -0.47, p<0.0001 | |
| Amyloid Beta PET Centiloid | N = 765 | N = 812 |
| Mean baseline | 104.0 | 101.8 |
| Adjusted mean change from baseline | -87.0 | -0.7 |
| Difference from placebo | -86.4, p<0.0001 | |
| Plasma p-tau217 (log10 transformed)a | N = 758 | N = 786 |
| Mean baseline | 0.67 | 0.66 |
| Adjusted mean change from baseline | -0.19 | 0.03 |
| Difference from placebo | -0.22, p<0.0001 | |
Exposure-Response Relationships
Model based exposure-response analyses for Study 1 demonstrated that exposures to donanemab-azbt were associated with a reduction in clinical decline on iADRS and CDR-SB. An association between reduction in amyloid beta plaque from baseline and clinical decline on iADRS and CDR-SB was also observed.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of KISUNLA were characterized using a population PK analysis with concentration data collected from 2131 patients with Alzheimer's disease who received KISUNLA in single or multiple doses. Accumulation of <1.3-fold occurs with every-4-week dosing; steady-state exposures are achieved after a single dose. In single doses from 10 to 40 mg/kg (~2 times the approved recommended dosage of 1400 mg for 70 kg of body weight), and multiple 10 and 20 mg/kg doses, exposures (Cmax and AUC) increased proportionally.
Elimination
KISUNLA is expected to be degraded by proteolytic enzymes in the same manner as endogenous IgG. The mean terminal half-life of donanemab-azbt is approximately 12.1 days. Donanemab-azbt clearance is 0.0255 L/h.
Specific Populations
Age, sex, or race were not found to affect the pharmacokinetics of donanemab-azbt. While body weight was found to influence both clearance and volume of distribution, the resulting changes were not clinically significant.
Patients with Renal or Hepatic Impairment
No clinical studies were conducted to evaluate the pharmacokinetics of donanemab-azbt in patients with renal or hepatic impairment. Donanemab-azbt is degraded by proteolytic enzymes and is not expected to undergo renal elimination or metabolism by hepatic enzymes.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADAs in other studies, including those of donanemab-azbt or of other donanemab products.
In up to 18 months of treatment in Study 1, 87% (691/792) of patients receiving KISUNLA once monthly developed anti-donanemab-azbt antibodies, and of those, 100% (691/691) had neutralizing antibodies.
Anti-donanemab-azbt antibody formation was associated with a higher incidence of infusion-related reactions compared to placebo [see Adverse Reactions (6.1)].
Anti-Drug Antibody Effects on Pharmacokinetics and Pharmacodynamics
The presence of anti-donanemab-azbt antibodies increased donanemab-azbt clearance. Among patients treated with KISUNLA in the placebo-controlled studies who developed anti-donanemab-azbt antibodies, mean donanemab-azbt serum trough concentrations at various time points were lower compared to patients who had not developed anti-donanemab-azbt antibodies. Patients with high ADA titers showed less reduction in amyloid plaque compared to patients with low ADA titers. However, there was no identified clinically significant effect of anti-donanemab-azbt antibodies on the effectiveness of KISUNLA over the treatment duration of 18 months.
14. Clinical Studies
The efficacy of KISUNLA was evaluated in a double-blind, placebo-controlled, parallel-group study (Study 1, NCT04437511) in patients with Alzheimer's disease (patients with confirmed presence of amyloid pathology and mild cognitive impairment or mild dementia stage of disease, consistent with Stage 3 and Stage 4 Alzheimer's disease). Patients were enrolled with a Mini-Mental State Examination (MMSE) score of ≥20 and ≤28 and had a progressive change in memory function for at least 6 months. Patients were included in the study based on visual assessment of tau PET imaging with flortaucipir and standardized uptake value ratio (SUVR). Patients were enrolled with or without concomitant approved therapies (cholinesterase inhibitors and the N-methyl-D-aspartate antagonist memantine) for Alzheimer's disease. Patients could enroll in an optional, long-term extension.
In Study 1, 1736 patients were randomized 1:1 to receive 700 mg of KISUNLA every 4 weeks for the first 3 doses, and then 1400 mg every 4 weeks (N = 860) or placebo (N = 876) for a total of up to 72 weeks. The treatment was switched to placebo based on amyloid PET levels measured at Week 24, Week 52, and Week 76. If the amyloid plaque level was <11 Centiloids on a single PET scan or 11 to <25 Centiloids on 2 consecutive PET scans, the patient was eligible to be switched to placebo.
Additionally, dose adjustments were allowed for treatment-emergent ARIA or symptoms that then showed ARIA-E or ARIA-H on MRI.
At baseline, mean age was 73 years, with a range of 59 to 86 years. Of the total number of patients randomized, 68% had low/medium tau level and 32% had high tau level; 71% were ApoE ε4 carriers and 29% were ApoE ε4 noncarriers. Fifty-seven percent of patients were female, 91% were White, 6% were Asian, 4% were Hispanic or Latino, and 2% were Black or African American.
The primary efficacy endpoint was change in the integrated Alzheimer's Disease Rating Scale (iADRS) score from baseline to 76 weeks. The iADRS is a combination of two scores: the Alzheimer's Disease Assessment Scale-Cognitive subscale (ADAS-Cog13) and the Alzheimer's Disease Cooperative Study – instrumental Activities of Daily Living (ADCS-iADL) scale. The total score ranges from 0 to 144, with lower scores reflecting worse cognitive and functional performance. Other efficacy endpoints included Clinical Dementia Rating Scale – Sum of Boxes (CDR-SB), ADAS-Cog13, and ADCS-iADL.
There were two primary analysis populations based on tau PET imaging with flortaucipir: 1) low/medium tau level population (defined by visual assessment and SUVR of ≥1.10 and ≤1.46), and 2) combined population of low/medium plus high tau (defined by visual assessment and SUVR >1.46) population.
Patients treated with KISUNLA demonstrated a statistically significant reduction in clinical decline on iADRS compared to placebo at Week 76 in the combined population (2.92, p<0.0001) and the low/medium tau population (3.25, p<0.0001).
Patients treated with KISUNLA demonstrated a statistically significant reduction in clinical decline on CDR-SB compared to placebo at Week 76 in the combined population (-0.70, p<0.0001) (see Figure 2 and Table 8). There were also statistically significant differences (p<0.001) between treatment groups as measured by ADAS-Cog13 and ADCS-iADL at Week 76 (see Table 8).
Dosing was continued or stopped in response to observed effects on amyloid imaging. The percentages of patients eligible for switch to placebo based on amyloid PET levels at Week 24, Week 52, and Week 76 timepoints were 17%, 47%, and 69%, respectively. Amyloid PET values may increase after treatment with donanemab is stopped [see Clinical Pharmacology (12.2)]. There is no data beyond the 76-week duration of Study 1 to guide whether additional dosing with KISUNLA may be needed for longer-term clinical benefit.
Figure 2: CDR-SB Change From Baseline in Combined Population Through 76 Weeks in Study 1a

a ****p<0.0001 versus placebo.
|
a Abbreviations: ADAS-Cog13 = Alzheimer's Disease Assessment Scale – 13-item Cognitive Subscale; ADCS-iADL = Alzheimer's Disease Cooperative Study – instrumental Activities of Daily Living subscale; CDR-SB = Clinical Dementia Rating Scale – Sum of Boxes; NCS2 = natural cubic spline with 2 degrees of freedom; MMRM = mixed model for repeated measures. |
||
|
b Assessed using MMRM analysis. |
||
|
c Assessed using NCS2 analysis. |
||
|
d Percent slowing of decline relative to placebo: difference of adjusted mean change from baseline between treatment groups divided by adjusted mean change from baseline of placebo group at Week 76. |
||
| Clinical Endpoints | KISUNLA
(N = 860) | Placebo
(N = 876) |
| CDR-SBb | ||
| Mean baseline | 3.92 | 3.89 |
| Adjusted mean change from baseline | 1.72 | 2.42 |
| Difference from placebo (%)d | -0.70 (29%) p<0.0001 | -- |
| ADAS-Cog13c | ||
| Mean baseline | 28.53 | 29.16 |
| Adjusted mean change from baseline | 5.46 | 6.79 |
| Difference from placebo (%)d | -1.33 (20%) p=0.0006 | -- |
| ADCS-iADLc | ||
| Mean baseline | 47.96 | 47.98 |
| Adjusted mean change from baseline | -4.42 | -6.13 |
| Difference from placebo (%)d | 1.70 (28%) p=0.0001 | -- |
16. How is Kisunla supplied
16.1 How Supplied
KISUNLA (donanemab-azbt) injection is a sterile, preservative-free, clear to opalescent, colorless to slightly yellow to slightly brown solution. KISUNLA is supplied in one vial per carton as follows:
350 mg/20 mL (17.5 mg/mL) single-dose vial: NDC 0002-9401-01.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Amyloid Related Imaging Abnormalities
Inform patients that KISUNLA may cause Amyloid Related Imaging Abnormalities or “ARIA”. ARIA most commonly presents as temporary swelling in areas of the brain that usually resolves over time. Some people may also have small spots of bleeding in or on the surface of the brain. Inform patients that most people with swelling in areas of the brain do not experience symptoms, however some people may experience symptoms such as headache, confusion, dizziness, vision changes, nausea, aphasia, weakness, or seizure. Instruct patients to notify their healthcare provider if these symptoms occur. Inform patients that events of intracerebral hemorrhage greater than 1 cm in diameter have been reported infrequently in patients taking KISUNLA, and that use of antithrombotic or thrombolytic medications while taking KISUNLA may increase the risk of bleeding in the brain. Notify patients that their healthcare provider will perform MRI scans to monitor for ARIA [see Warnings and Precautions (5.1)].
Inform patients that although ARIA can occur in any patient treated with KISUNLA, there is an increased risk in patients who are ApoE ε4 homozygotes, and that testing for ApoE ε4 status should be performed prior to initiation of treatment to inform the risk of developing ARIA. Prior to testing, discuss with patients the risk of ARIA across genotypes and the implications of genetic testing results. Inform patients that if testing is not performed, it cannot be determined if they are ApoE ε4 homozygotes and at a higher risk for ARIA.
Inform patients that some symptoms of ARIA can mimic ischemic stroke and that their healthcare providers may need to perform additional testing to determine how to treat those symptoms in patients taking KISUNLA. Advise patients to carry information that they are being treated with KISUNLA.
Patient Registry
Providers should encourage patients to participate in real world data collection (e.g., registries) to help further the understanding of Alzheimer's disease and the impact of Alzheimer's disease treatments. Providers and patients can contact 1-800-LillyRx (1-800-545-5979) for a list of currently enrolling programs.
Hypersensitivity Reactions
Inform patients that KISUNLA may cause hypersensitivity reactions, including anaphylaxis and angioedema, and to contact their healthcare provider if hypersensitivity reactions occur [see Warnings and Precautions (5.2)].
Infusion-Related Reactions
Inform patients that KISUNLA may cause infusion-related reactions, including chills, erythema, nausea, vomiting, difficulty breathing, sweating, headache, chest pain, and high or low blood pressure, and to contact their healthcare provider if infusion-related reactions occur [see Warnings and Precautions (5.3)].
Eli Lilly and Company, Indianapolis, IN 46285, USA
US License No. 1891
KIS-0001-USPI-20240702
|
This Medication Guide has been approved by the U.S. Food and Drug Administration |
Issued: 7/2024 |
||||
| MEDICATION GUIDE
KISUNLA™ (kih-SUHN-lah) (donanemab-azbt) injection, for intravenous use |
|||||
| What is the most important information I should know about KISUNLA?
KISUNLA can cause serious side effects, including: Amyloid Related Imaging Abnormalities or ARIA. ARIA is a common side effect that does not usually cause any symptoms, but serious symptoms can occur. ARIA can be fatal. It is most commonly seen as temporary swelling in areas of the brain that usually resolves over time. Some people may also have small spots of bleeding in or on the surface of the brain, and infrequently, larger areas of bleeding in the brain can occur. Most people who develop ARIA do not get symptoms; however, some people may have symptoms such as: |
|||||
|
|
|
|
||
| Some people have a genetic risk factor (homozygous apolipoprotein E ε4 gene carriers) that may cause an increased risk for ARIA. Talk to your healthcare provider about testing to see if you have this risk factor. You may be at a higher risk of developing bleeding in the brain if you take medicines to reduce blood clots from forming (antithrombotic medicines) while receiving KISUNLA. Your healthcare provider will do magnetic resonance imaging (MRI) scans before and during your treatment with KISUNLA to check you for ARIA. You should carry information that you are receiving KISUNLA which can cause ARIA, and that ARIA symptoms can look like stroke symptoms. Call your healthcare provider or go to the nearest hospital emergency room right away if you have any of the symptoms listed above. There are registries that collect information on treatments for Alzheimer's disease. Your healthcare provider can help you become enrolled in these registries. For more information, go to www.kisunla.com or call 1-800-LillyRx (1-800-545-5979). |
|||||
What is KISUNLA?
|
|||||
| It is not known if KISUNLA is safe and effective in children. | |||||
Do not receive KISUNLA if you:
|
|||||
Before receiving KISUNLA, tell your healthcare provider about all of your medical conditions, including if you:
|
|||||
| Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Especially tell your healthcare provider if you take medicines to reduce blood clots from forming (antithrombotic medicines, including aspirin). Ask your healthcare provider for a list of these medications if you are not sure. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|||||
How will I receive KISUNLA?
|
|||||
| What are the possible side effects of KISUNLA?
KISUNLA can cause serious side effects, including:
|
|||||
| If you have ever had an infusion-related reaction while receiving KISUNLA, your healthcare provider may give you medicines before your KISUNLA infusions to decrease your chance of having an infusion reaction. These medicines may include an antihistamine, acetaminophen, or a steroid. The most common side effects of KISUNLA include:
|
|||||
| These are not all the possible side effects of KISUNLA. For more information ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||||
| General information about the safe and effective use of KISUNLA
Medicines are sometimes prescribed for purposes other than those listed in this Medication Guide. You can ask your pharmacist or healthcare provider for more information about KISUNLA that is written for health professionals. |
|||||
| What are the ingredients in KISUNLA?
Active ingredient: donanemab-azbt Inactive ingredients: anhydrous citric acid, polysorbate 80, sodium citrate, sucrose, and Water for Injection, USP KISUNLA is a trademark of Eli Lilly and Company. For more information, go to www.kisunla.com or call 1-800-LillyRx (1-800-545-5979). Eli Lilly and Company, Indianapolis, IN 46285, USA US License No. 1891 Copyright © 2024, Eli Lilly and Company. All rights reserved. |
|||||
KIS-0001-MG-20240702
| KISUNLA
donanemab-azbt injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Eli Lilly and Company (006421325) |
| Registrant - Eli Lilly and Company (006421325) |
Biological Products Related to Kisunla
Find detailed information on biosimilars for this medication.
Frequently asked questions
- How do I decide between Leqembi and Kisunla?
- Will insurance cover Kisunla treatment?
- How well does Kisunla work?
- How long will I need to take Kisunla?
- How quickly does Kisunla start working?
- Can Kisunla be used with other Alzheimer's drugs?
More about Kisunla (donanemab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: miscellaneous central nervous system agents
- En español
Patient resources
Professional resources
Related treatment guides
Copyright © 2024, Eli Lilly and Company. All rights reserved.