Kcentra: Package Insert / Prescribing Info
Package insert / product label
Generic name: prothrombin, coagulation factor vii human, coagulation factor ix human, coagulation factor x human, protein c, protein s human
Dosage form: injection
Drug class: Anticoagulant reversal agents
J Code (medical billing code): J7168 (Per intl units, injection)
Medically reviewed by Drugs.com. Last updated on May 21, 2023.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
KCENTRA (Prothrombin Complex Concentrate (Human))
For Intravenous Use, Lyophilized Powder for Reconstitution
Initial U.S. Approval: 2013
WARNING: ARTERIAL AND VENOUS THROMBOEMBOLIC COMPLICATIONS
Patients being treated with Vitamin K antagonists (VKA) therapy have underlying disease states that predispose them to thromboembolic events. Potential benefits of reversing VKA should be weighed against the potential risks of thromboembolic events, especially in patients with the history of a thromboembolic event. Resumption of anticoagulation should be carefully considered as soon as the risk of thromboembolic events outweighs the risk of acute bleeding.
- Both fatal and non-fatal arterial and venous thromboembolic complications have been reported with Kcentra in clinical trials and post marketing surveillance. Monitor patients receiving KCENTRA for signs and symptoms of thromboembolic events.
- KCENTRA was not studied in subjects who had a thromboembolic event, myocardial infarction, disseminated intravascular coagulation, cerebral vascular accident, transient ischemic attack, unstable angina pectoris, or severe peripheral vascular disease within the prior 3 months. KCENTRA may not be suitable in patients with thromboembolic events in the prior 3 months. (5.2)
Recent Major Changes
| Warnings and Precautions (5.2) | 05/2023 |
Indications and Usage for Kcentra
KCENTRA, Prothrombin Complex Concentrate (Human), is a blood coagulation factor replacement product indicated for the urgent reversal of acquired coagulation factor deficiency induced by Vitamin K antagonist (VKA, e.g., warfarin) therapy in adult patients with:
- acute major bleeding or
- need for an urgent surgery/invasive procedure. (1)
Kcentra Dosage and Administration
For intravenous use after reconstitution only.
- KCENTRA dosing should be individualized based on the patient's baseline International Normalized Ratio (INR) value, and body weight. (2.1)
- Administer Vitamin K concurrently to patients receiving KCENTRA to maintain factor levels once the effects of KCENTRA have diminished.
- The safety and effectiveness of repeat dosing have not been established and it is not recommended. (2.1)
- Administer reconstituted KCENTRA at a rate of 0.12 mL/kg/min (~3 units/kg/min) up to a maximum rate of 8.4 mL/min (~210 units/min). (2.3)
| Pre-treatment INR | 2–< 4 | 4–6 | > 6 |
|---|---|---|---|
|
|||
| Dose* of KCENTRA (units† of Factor IX) / kg body weight | 25 | 35 | 50 |
| Maximum dose‡ (units of Factor IX) | Not to exceed 2500 | Not to exceed 3500 | Not to exceed 5000 |
Dosage Forms and Strengths
KCENTRA is available as a white or slightly colored lyophilized concentrate in a single-dose vial containing coagulation Factors II, VII, IX and X, and antithrombotic Proteins C and S. (3)
Contraindications
KCENTRA is contraindicated in patients with:
- Known anaphylactic or severe systemic reactions to KCENTRA or any components in KCENTRA including heparin, Factors II, VII, IX, X, Proteins C and S, Antithrombin III and human albumin. (4)
- Disseminated intravascular coagulation. (4)
- Known heparin-induced thrombocytopenia. KCENTRA contains heparin. (4)
Warnings and Precautions
- Hypersensitivity reactions may occur. If necessary, discontinue administration and institute appropriate treatment. (5.1)
- Arterial and venous thromboembolic complications have been reported in patients receiving KCENTRA. Monitor patients receiving KCENTRA for signs and symptoms of thromboembolic events. KCENTRA was not studied in subjects who had a thrombotic or thromboembolic (TE) event within the prior 3 months. KCENTRA may not be suitable in patients with thromboembolic events in the prior 3 months. (5.2)
- KCENTRA is made from human blood and may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent, and theoretically, the Creutzfeldt-Jakob disease (CJD) agent. (5.3)
Adverse Reactions/Side Effects
- The most common adverse reactions (ARs) (frequency ≥ 2.8%) observed in subjects receiving KCENTRA were headache, nausea/vomiting, hypotension, and anemia. (6)
- The most serious ARs were thromboembolic events including stroke, pulmonary embolism, and deep vein thrombosis. (6)
To report SUSPECTED ADVERSE REACTIONS, contact CSL Behring at 1-866-915-6958 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2023
Full Prescribing Information
WARNING: ARTERIAL AND VENOUS THROMBOEMBOLIC COMPLICATIONS
Patients being treated with Vitamin K antagonists (VKA) therapy have underlying disease states that predispose them to thromboembolic events. Potential benefits of reversing VKA should be weighed against the potential risks of thromboembolic events (TE), especially in patients with the history of a thromboembolic event. Resumption of anticoagulation should be carefully considered as soon as the risk of thromboembolic events outweighs the risk of acute bleeding.
- Both fatal and non-fatal arterial and venous thromboembolic complications have been reported with KCENTRA in clinical trials and post marketing surveillance. Monitor patients receiving KCENTRA for signs and symptoms of thromboembolic events. (5.2)
- KCENTRA was not studied in subjects who had a thromboembolic event, myocardial infarction, disseminated intravascular coagulation, cerebral vascular accident, transient ischemic attack, unstable angina pectoris, or severe peripheral vascular disease within the prior 3 months. KCENTRA may not be suitable in patients with thromboembolic events in the prior 3 months. (5.2)
1. Indications and Usage for Kcentra
KCENTRA, (Prothrombin Complex Concentrate (Human)), is a blood coagulation factor replacement product indicated for the urgent reversal of acquired coagulation factor deficiency induced by Vitamin K antagonist (VKA, e.g., warfarin) therapy in adult patients with:
- acute major bleeding or
- need for an urgent surgery/invasive procedure.
2. Kcentra Dosage and Administration
For intravenous use after reconstitution only.
2.1 Dosage
- Measurement of INR prior to treatment and close to the time of dosing is important because coagulation factors may be unstable in patients with acute major bleeding or an urgent need for surgery and other invasive procedures.
- Individualize KCENTRA dosing based on the patient's current pre-dose International Normalized Ratio (INR) value, and body weight (see Table 1).
- The actual potency per vial of Factors II, VII, IX and X, Proteins C and S is stated on the carton.
- Administer Vitamin K concurrently to patients receiving KCENTRA. Vitamin K is administered to maintain Vitamin K-dependent clotting factor levels once the effects of KCENTRA have diminished.
- The safety and effectiveness of repeat dosing have not been established and it is not recommended.
- Dose ranging within pre-treatment INR groups has not been studied in randomized clinical trials of KCENTRA.
| Pre-treatment INR | 2–< 4 | 4–6 | > 6 |
|---|---|---|---|
|
|||
| Dose* of KCENTRA (units† of Factor IX) / kg body weight | 25 | 35 | 50 |
| Maximum dose‡ (units of Factor IX) | Not to exceed 2500 | Not to exceed 3500 | Not to exceed 5000 |
Example dosing calculation for 80 kg patient
For example, an 80 kg patient with a baseline of INR of 5.0, the dose would be 2,800 Factor IX units of KCENTRA, calculated as follows based on INR range of 4–6, see Table 1:
35 units of Factor IX/kg × 80 kg = 2,800 units of Factor IX required1
Monitor INR and clinical response during and after treatment. In clinical trials, KCENTRA decreased the INR to ≤ 1.3 within 30 minutes in most subjects. The relationship between this or other INR values and clinical hemostasis in patients has not been established [see Clinical Studies (14)].
- 1
- For a vial with an actual potency of 30 units/mL Factor IX, 93 mL would be given (2,800 U/30 U per mL = 93 mL).
2.2 Preparation and Reconstitution
- Reconstitute KCENTRA using aseptic technique with 20 mL (nominal potency 500 U kit) or 40 mL (nominal potency 1000 U kit) of Sterile Water for Injection (diluent) provided in the kit.
- Do not use KCENTRA beyond the expiration date on the vial label and carton.
- KCENTRA is for single-dose only. Contains no preservatives. Discard partially used vials.
Kcentra Reconstitution Instructions
- Ensure that the KCENTRA vial and diluent vial are at room temperature.
- Remove flip caps from the KCENTRA and diluent vials.
- Wipe the stoppers with the alcohol swab provided and allow to dry prior to opening the Mix2Vial package.
- Open the Mix2Vial package by peeling off the lid (Fig. 1). Do not remove the Mix2Vial from the blister package.

Fig. 1
- Place the diluent vial on an even, clean surface and hold the vial tight. Take the Mix2Vial together with the blister package and push the spike of the blue adapter end straight down through the diluent vial stopper (Fig. 2).

Fig. 2
- Carefully remove the blister package from the Mix2Vial set by holding at the rim, and pulling vertically upwards. Make sure that you only pull away the blister package and not the Mix2Vial set (Fig. 3).

Fig. 3
- Place the KCENTRA vial on an even and firm surface. Invert the diluent vial with the Mix2Vial set attached and push the spike of the transparent adapter end straight down through the KCENTRA vial stopper (Fig. 4). The diluent will automatically flow into the KCENTRA vial.

Fig. 4
Note: If the vacuum in the KCENTRA vial is accidentally lost during reconstitution with the Mix2Vial device, the transfer with the Mix2Vial will not work.
In this case, separate the set into two pieces as illustrated in Fig. 6 below; do not discard the diluent vial. Place the KCENTRA vial aside on a flat surface. Remove the blue adapter end from the diluent vial of the Mix2Vial set (Fig. 5) by lifting and bending the blue adapter to the side until it disconnects from the diluent vial.
For reconstitution:- Using a separate sterile needle and syringe, withdraw the remaining diluent. Remove the needle from the syringe.
- Attach the syringe to the transparent adapter of the KCENTRA vial as illustrated in Fig. 8 below, and transfer the entire diluent volume into the KCENTRA vial. Remove syringe.
- Gently swirl the KCENTRA vial to ensure the product is fully dissolved. Do not shake.
- Proceed to step 10.
Fig. 5
- With one hand, grasp the KCENTRA-side of the Mix2Vial set and with the other hand grasp the diluent-side and unscrew the set carefully counterclockwise into two pieces (Fig. 6). Discard the diluent vial with the blue Mix2Vial adapter attached.

Fig. 6
- Gently swirl the KCENTRA vial with the transparent adapter attached until the substance is fully dissolved (Fig. 7). Do not shake.

Fig. 7
- Draw air into an empty, sterile syringe. While the KCENTRA vial is upright, connect the syringe to the Mix2Vial's Luer Lock fitting by screwing clockwise. Inject air into the KCENTRA vial (Fig. 8).

Fig. 8
- While keeping the syringe plunger pressed, turn the system upside down and draw the solution into the syringe by pulling the plunger back slowly (Fig. 9).

Fig. 9
- Now that the solution has been transferred into the syringe, firmly hold on to the barrel of the syringe (keeping the syringe plunger facing down) and disconnect the transparent Mix2Vial adapter from the syringe by unscrewing counterclockwise (Fig. 10). Attach the syringe to a suitable intravenous administration set.

Fig. 10
- After reconstitution, administration should begin promptly or within 4 hours.
- If the same patient is to receive more than one vial, you may pool the contents of multiple vials. Use a separate unused Mix2Vial transfer set for each product vial.
2.3 Administration
- Do not mix KCENTRA with other medicinal products; administer through a separate infusion line.
- Visually inspect the reconstituted solution for particulate matter and discoloration prior to administration whenever solution and container permit. Reconstituted KCENTRA solution should be colorless, clear to slightly opalescent, and free from visible particles. Do not use if the solution is cloudy, discolored, or contains particulates.
- Use aseptic technique when administering KCENTRA.
- Administer at room temperature.
- Administer by intravenous infusion at a rate of 0.12 mL/kg/min (~3 units/kg/min), up to a maximum rate of 8.4 mL/min (~210 units/min).
- No blood should enter the syringe, as there is a possibility of fibrin clot formation.
3. Dosage Forms and Strengths
- KCENTRA is available as a white or slightly colored lyophilized concentrate in a single-dose vial containing coagulation Factors II, VII, IX and X, and antithrombotic Proteins C and S.
- KCENTRA potency (units) is defined by Factor IX content. The actual potency for 500 unit vial ranges from 400-620 Factor IX units/vial. The actual potency for 1000 unit vial ranges from 800-1240 Factor IX units/vial. The actual content of Factor IX as measured in units of potency for the vial before reconstitution is stated by the expiration date. When reconstituted, the final concentration of drug product in Factor IX units will be in a range from 20–31 units/mL.
- The actual units of potency for each coagulation factor (Factors II, VII, IX and X), and Proteins C and S are stated on the carton.
4. Contraindications
KCENTRA is contraindicated in:
- Patients with known anaphylactic or severe systemic reactions to KCENTRA or any components in KCENTRA including heparin, Factors II, VII, IX, X, Proteins C and S, Antithrombin III and human albumin.
- Patients with disseminated intravascular coagulation (DIC).
- Patients with known heparin-induced thrombocytopenia (HIT). KCENTRA contains heparin [see Description (11)].
5. Warnings and Precautions
5.1 Hypersensitivity Reactions
Hypersensitivity reactions including flushing, urticaria, tachycardia, anxiety, angioedema, wheezing, nausea, vomiting, hypotension, tachypnea, dyspnea, pulmonary edema, and bronchospasm have been observed with KCENTRA.
If severe allergic reaction or anaphylactic type reactions occur, immediately discontinue administration, and institute appropriate treatment.
5.2 Thromboembolic Risk/Complications
Both fatal and non-fatal arterial thromboembolic events (including acute myocardial infarction and arterial thrombosis), and venous thromboembolic events (including pulmonary embolism and venous thrombosis) and disseminated intravascular coagulation have been reported with KCENTRA in clinical trials and post marketing surveillance [see Adverse Reactions (6) and Clinical Studies (14)]. Patients being treated with VKA therapy have underlying disease states that predispose them to thromboembolic events. Reversing VKA therapy exposes patients to the thromboembolic risk of their underlying disease. Resumption of anticoagulation should be carefully considered following administration of KCENTRA and Vitamin K once the risk of thromboembolic events outweighs the risk of bleeding.
Thromboembolic events occurred more frequently following KCENTRA compared to plasma in a randomized, plasma controlled trial in subjects requiring urgent reversal of VKA anticoagulation due to acute major bleeding, and the excess in thromboembolic events was more pronounced among subjects who had a history of prior thromboembolic event, although these differences were not statistically significant [see Adverse Reactions (6.1) and Clinical Studies (14)].
In a postmarketing observational study, there was no significant difference in the risk of post-treatment thromboembolic events after receiving KCENTRA compared with plasma therapy for VKA-associated acute major bleeding [see Adverse Reactions (6.2)].
Potential benefits of treatment with KCENTRA should be weighed against the potential risks of thromboembolic events [see Adverse Reactions (6)]. Patients with a history of thrombotic events, myocardial infarction, cerebral vascular accident, transient ischemic attack, unstable angina pectoris, severe peripheral vascular disease, or disseminated intravascular coagulation, within the previous 3 months were excluded from participating in the plasma-controlled RCT. KCENTRA may not be suitable in patients with thromboembolic events in the prior 3 months. Because of the risk of thromboembolism associated with reversal of VKA, closely monitor patients for signs and symptoms of thromboembolism during and after administration of KCENTRA [see 17 Patient Counseling Information].
5.3 Transmissible Infectious Agents
Because KCENTRA is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent, and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. There is also the possibility that unknown infectious agents may be present in such products. Despite the use of two dedicated virus reduction steps in manufacturing to reduce risks, such products may still potentially transmit disease.
Reports of suspected virus transmission of hepatitis A, B, C, and HIV were generally confounded by concomitant administration of blood/blood components and/or other plasma-derived products. No causal relationship to KCENTRA administration was established for any of these reports since introduction of a virus filtration step in 1996.
All infections thought by a physician to have been possibly transmitted by KCENTRA should be reported by the physician or other healthcare provider to the CSL Behring Pharmacovigilance Department at 1-866-915-6958 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6. Adverse Reactions/Side Effects
The most common adverse reactions (ARs) (frequency ≥2.8%) observed in subjects receiving KCENTRA were headache, nausea/vomiting, hypotension, and anemia.
The most serious ARs were thromboembolic events including stroke, pulmonary embolism, and deep vein thrombosis.
The following serious adverse reactions are described below and/or elsewhere in the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Arterial and venous thromboembolic complications [see Boxed Warning and Warnings and Precautions (5.2)]
- Possible transmission of infectious agents [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Randomized, Plasma-Controlled Trial in Acute Major Bleeding
In a prospective, randomized, open-label, active-controlled multicenter non-inferiority trial, 212 subjects who required urgent reversal of VKA therapy due to acute major bleeding were enrolled and randomized to treatment; 103 were treated with KCENTRA and 109 with plasma. Subjects with a history of a thrombotic event, myocardial infarction, cerebral vascular accident, transient ischemic attack, unstable angina pectoris, severe peripheral vascular disease, or disseminated intravascular coagulation, within the previous 3 months were excluded from participating. Subjects ranged in age from 26 years to 96 years.
Randomized, Plasma-Controlled Trial in Urgent Surgery/Invasive Procedures
In a prospective, randomized, open-label, active-controlled, multicenter non-inferiority trial, 176 subjects who required urgent reversal of VKA therapy due to the need for an urgent surgical or urgent invasive procedure were enrolled; 88 were treated with KCENTRA and 88 with plasma. Subjects ranged in age from 27 years to 94 years.
Adverse reactions are summarized for KCENTRA and plasma in the Acute Major Bleeding and Urgent Surgery/Invasive Procedures RCTs (see Table 2).
Adverse Reactions are defined as adverse events that began during or within 72 hours of test product infusion plus adverse events considered possibly/probably related or related to study treatment according to the investigator, sponsor, or the blinded safety adjudication board (SAB), and with at least a 1.3‐fold difference between treatments.
| No. (%) of subjects | ||
|---|---|---|
| KCENTRA (N = 191) | Plasma (N = 197) |
|
| Nervous system disorders | ||
| Headache | 14 (7.3%) | 7 (3.6%) |
| Respiratory, thoracic, and mediastinal disorders | ||
| Pleural effusion | 8 (4.2%) | 3 (1.5%) |
| Respiratory distress/dyspnea/hypoxia | 7 (3.7%) | 10 (5.1%) |
| Pulmonary edema | 3 (1.6%) | 10 (5.1%) |
| Gastrointestinal disorders | ||
| Nausea/vomiting | 12 (6.3%) | 8 (4.1%) |
| Diarrhea | 4 (2.1%) | 7 (3.6%) |
| Cardiac disorders | ||
| Tachycardia | 9 (4.7%) | 2 (1.0%) |
| Atrial fibrillation | 8 (4.2%) | 6 (3.0%) |
| Metabolism and nutrition disorders | ||
| Fluid overload* | 5 (2.6%) | 16 (8.1%) |
| Hypokalemia | 9 (4.7%) | 14 (7.1%) |
| Psychiatric disorders | ||
| Insomnia | 9 (4.7%) | 6 (3.0%) |
| Vascular disorders | ||
| Hypotension† | 14 (7.3%) | 10 (5.1%) |
| Injury, poisoning, and procedural complications | ||
| Skin laceration/contusion/subcutaneous hematoma | 8 (4.2%) | 5 (2.5%) |
| Blood and lymphatic disorders | ||
| Anemia‡ | 11 (5.8%) | 16 (8.1%) |
Serious adverse reactions in subjects receiving KCENTRA in both RCTs included ischemic cerebrovascular accident (stroke), DVT, thrombosis, and venous insufficiency. Serious adverse reactions in both RCTs for plasma included myocardial ischemia, myocardial infarction, fluid overload, embolic cerebral infarction, pulmonary edema, respiratory failure, and DVT.
There were a total of 10 subjects (9.7%) who died in the KCENTRA group (1 additional death occurred on day 46 just after completion of the study reporting period) and 5 (4.6%) who died in the plasma group in the plasma-controlled RCT in acute major bleeding. The 95% confidence interval for the KCENTRA minus plasma between-group difference in deaths ranged from -2.7% to 13.5%. From the plasma-controlled RCT in urgent surgery/invasive procedures, there were a total of 3 subjects (3.4%) who died in the KCENTRA group (1 additional death occurred on day 48 after completion of the study reporting period) and 8 (9.1%) who died in the plasma group. The 95% confidence interval for the KCENTRA minus plasma between-group difference in deaths in this trial ranged from -14.6% to 2.7%. One death in the KCENTRA group in the RCT in Acute Major Bleeding and one death in the plasma group in the RCT in urgent surgery/invasive procedures were considered possibly related to study treatment according to an assessment of masked data by an independent safety adjudication board. No factors common to all deaths were identified, except for the frequent findings of a high comorbidity burden, advanced age, and death after being placed on comfort care. Although, a greater proportion of subjects in the RCT in acute major bleeding than in the RCT in surgery/invasive procedure received the highest two recommended doses of KCENTRA because more subjects in the trial in acute major bleeding had a baseline INR in the ranges of 4–6 and > 6.0, an analysis of deaths and factor levels in subjects with major bleeding revealed that subjects who died had similar median factor levels to subjects that did not die. Additionally, outliers with supraphysiologic factor levels did not have a mortality rate out of proportion to the overall population.
Fluid Overload
There were 9 subjects (4.7%, all non-related by investigator assessment) in the KCENTRA group who experienced fluid overload in the plasma-controlled RCTs in acute major bleeding and urgent surgery/invasive procedures and 25 (12.7%, 13 events related by investigator assessment) who had fluid overload in the plasma group. The 95% confidence interval for the KCENTRA minus Plasma between-group difference in fluid overload event incidence ranged from -14.1% to -2.0%.
Subgroup analyses of the RCTs in acute major bleeding and urgent surgery/invasive procedures according to whether subjects with fluid overload events had a prior history of congestive heart failure are presented in Table 3.
| Subgroup | Acute Major Bleeding Study | Urgent Surgery/Invasive Procedures Study | ||||||
|---|---|---|---|---|---|---|---|---|
| KCENTRA | Plasma | KCENTRA | Plasma | |||||
| N | Fluid Overload N (%) | N | Fluid Overload N (%) | N | Fluid Overload N (%) | N | Fluid Overload N (%) |
|
| All subjects | 103 | 6 (5.8) | 109 | 14 (12.8) | 88 | 3 (3.4) | 88 | 11 (12.5) |
| With history of CHF | 46 | 4 (8.7) | 44 | 11 (25.0) | 24 | 1 (4.2) | 36 | 6 (16.7) |
| Without history of CHF | 57 | 2 (3.5) | 65 | 3 (4.6) | 64 | 2 (3.1) | 52 | 5 (9.6) |
Thromboembolic Events
In RCTs, there were 13 subjects (6.8%) in the KCENTRA group who experienced possible thromboembolic events (TEEs) and 14 (7.1%) who had TEEs in the plasma group. The incidence of thromboembolic (TE) adverse reactions assessed as at least possibly related to study treatment by the Investigator or, in the case of serious thromboembolic events, the blinded safety adjudication board (SAB) was 9 (4.7%) in the KCENTRA group and 7 (3.6%) in the plasma group. When also considering the events which began during or within 72 hours of test product infusion, the incidence was 9 (4.7%) in the KCENTRA group and 8 (4.1%) in the plasma group.
TE events observed in the acute major bleeding and the urgent surgery/invasive procedures RCTs are shown in Table 4.
| No. (%) of subjects | ||||
|---|---|---|---|---|
| System Organ Class | Acute Major Bleeding Study | Urgent Surgery/Invasive Procedures Study | ||
| KCENTRA (N = 103) | Plasma (N = 109) | KCENTRA (N = 88) | Plasma (N = 88) |
|
|
||||
| Any possible TEE* | 9 (8.7%) | 6 (5.5%) | 4 (4.5) | 8 (9.1) |
| TEE Adverse reactions | 6 (5.5%) | 4 (3.7%) | 4 (4.5) | 4 (4.5) |
| Cardiac disorders | ||||
| Myocardial infarction | 0 | 1 (0.9%) | 0 | 2 (2.3) |
| Myocardial ischemia | 0 | 2 (1.8%) | 0 | 0 |
| Nervous system disorders | ||||
| Ischemic cerebrovascular accident (stroke) | 2 (1.9%) | 0 | 1 (1.1) | 0 |
| Embolic cerebral infarction | 0 | 0 | 0 | 1 (1.1) |
| Cerebrovascular disorder | 0 | 1 (0.9%) | 0 | 0 |
| Vascular disorders | ||||
| Venous thrombosis calf | 1 (1.0%) | 0 | 0 | 0 |
| Venous thrombosis radial vein | 0 | 0 | 1 (1.1) | 0 |
| Thrombosis (microthrombosis of toes) | 0 | 0 | 1 (1.1) | 0 |
| Deep vein thrombosis (DVT) | 1 (1.0%) | 0 | 1 (1.1) | 1 (1.1) |
| Fistula Clot | 1 (1.0%) | 0 | 0 | 0 |
| Unknown Cause of Death (not confirmed TEE) | ||||
| Sudden death | 1 (1.0%) | 0 | 0 | 0 |
Subgroup analyses of the RCTs according to whether subjects with thromboembolic events had a prior history of a thromboembolic event are presented in Table 5.
| Acute Major Bleeding Study | Urgent Surgery/Invasive Procedures Study | |||||||
|---|---|---|---|---|---|---|---|---|
| KCENTRA | Plasma | KCENTRA | Plasma | |||||
| N | TE Events*
N (%) | N | TE Events N (%) | N | TE Events*
N (%) | N | TE Events N (%) |
|
|
||||||||
| All subjects | 103 | 9 (8.7) | 109 | 6 (5.5) | 88 | 4 (4.5) | 88 | 8 (9.1) |
| With history of TE event† | 69 | 8 (11.6) | 79 | 3 (3.8) | 55 | 3 (5.5) | 62 | 5 (8.1) |
| Without history of TE event | 34 | 1 (2.9) | 30 | 3 (10.0) | 33 | 1 (3.0) | 26 | 3 (11.5) |
The European Bleeding and Surgical Study
In a prospective, open label, single-arm, multicenter safety and efficacy trial, 17 subjects who required urgent reversal of VKA due to acute bleeding were enrolled and 26 subjects who required urgent reversal of Vitamin K antagonist due to the need for an urgent surgical/invasive procedure were enrolled, all were treated with KCENTRA. Subjects ranged in age from 22 years to 85 years. Serious adverse reactions considered possibly related to KCENTRA included a suspected pulmonary embolism which occurred in one subject following a second dose of KCENTRA. A single non-fatal TE event occurred in another KCENTRA-treated subject in that trial.
6.2 Postmarketing Experience
In a postmarketing observational study, the risk of TEEs was assessed using Kaiser Permanente's databases in a matched, real-world cohort of hospitalized adults receiving either KCENTRA or plasma for VKA-associated major bleeding. For the primary analysis, 1119 KCENTRA patients and 1119 historical plasma patients with no recent history of TEEs were grouped in 1:1 individually matched cohort based on propensity scores. During the 45-day follow-up after acute VKA reversal, there was no statistically significant difference in the adjusted risk of TEEs between KCENTRA-treated patients and plasma-treated patients. . In a secondary analysis, 101 KCENTRA patients and 101 historical plasma patients with a recent history of TEEs diagnosed ≤90 days before study entry were also grouped in 1:1 individually matched cohort based on propensity scores. During the 45-day follow-up after acute VKA reversal, there was no statistically significant difference in the adjusted risk of TEEs between KCENTRA-treated patients and plasma-treated patients.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no data with KCENTRA use in pregnancy to inform on drug-associated risk. Animal reproduction studies have not been conducted with KCENTRA. It is not known whether KCENTRA can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. KCENTRA should be prescribed for a pregnant woman only if clearly needed.
In the U.S. general population, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.4 Pediatric Use
The safety and efficacy of KCENTRA in the pediatric population has not been studied.
8.5 Geriatric Use
Of the total number of subjects (431) with acute major bleeding or with the need for an urgent surgery/invasive procedure treated to reverse VKA anticoagulation in three clinical studies, 66% were 65 years old or greater and 39% were 75 years old or greater. There were no clinically significant differences between the safety profile of KCENTRA and plasma in any age group.
11. Kcentra Description
KCENTRA is a purified, heat-treated, nanofiltered and lyophilized non-activated four-factor Prothrombin Complex Concentrate (Human) prepared from human U.S. Source Plasma (21 CFR 640.60). It contains the Vitamin K dependent Coagulation Factors II, VII, IX and X, and the antithrombotic Proteins C and S. Factor IX is the lead factor for the potency of the preparation as stated on the vial label. The excipients are human antithrombin III, heparin, human albumin, sodium chloride, and sodium citrate. KCENTRA is sterile, pyrogen-free, and does not contain preservatives.
The product contents are shown in Table 6 and listed as ranges for the blood coagulation factors.
| Ingredient | Potency Range for 500 units | Potency Range for 1000 units |
|---|---|---|
|
||
| Total protein | 120–280 mg | 240–560 mg |
| Factor II | 380–800 units | 760–1600 units |
| Factor VII | 200–500 units | 400–1000 units |
| Factor IX | 400–620 units | 800–1240 units |
| Factor X | 500–1020 units | 1000–2040 units |
| Protein C | 420–820 units | 840–1640 units |
| Protein S | 240–680 units | 480–1360 units |
| Heparin | 8–40 units | 16–80 units |
| Antithrombin III | 4–30 units | 8–60 units |
| Human albumin | 40–80 mg | 80–160 mg |
| Sodium chloride | 60–120 mg | 120–240 mg |
| Sodium citrate | 40–80 mg | 80–160 mg |
| HCl | Small amounts | Small amounts |
| NaOH | Small amounts | Small amounts |
All plasma used in the manufacture of KCENTRA is obtained from US donors and is tested using serological assays for hepatitis B surface antigen and antibodies to HIV-1/2 and HCV. The plasma is tested with Nucleic Acid Testing (NAT) for HCV, HIV-1, HAV, and HBV, and found to be non-reactive (negative), and the plasma is also tested by NAT for human parvovirus B19 (B19V) in order to exclude donations with high titers. The limit for B19V in the fractionation pool is set not to exceed 104 units of B19V DNA per mL. Only plasma that passed virus screening is used for production.
The KCENTRA manufacturing process includes various steps, which contribute towards the reduction/inactivation of viruses. KCENTRA is manufactured from cryo-depleted plasma that is adsorbed via ion exchange chromatography, heat treated in aqueous solution for 10 hours at 60°C, precipitated, adsorbed to calcium phosphate, virus filtered, and lyophilized.
Manufacturing steps were independently validated in a series of in vitro experiments for their virus inactivation / reduction capacity for both enveloped and non-enveloped viruses. Table 7 shows the virus clearance during the manufacturing process for KCENTRA, expressed as the mean log10 reduction factor.
| Virus Studied | Manufacturing Steps | |||
|---|---|---|---|---|
| Heat treatment ("Pasteurization") | Ammonium sulphate precipitation followed by Ca Phosphate adsorption | 2 × 20 nm Virus Filtration | Overall Virus Reduction [log10] |
|
| HIV Human immunodeficiency virus, a model for HIV-1 and HIV-2 | ||||
| BVDV Bovine viral diarrhea virus, model for HCV | ||||
| PRV Pseudorabies virus, a model for large enveloped DNA viruses | ||||
| WNV West Nile virus | ||||
| HAV Hepatitis A virus | ||||
| CPV Canine parvovirus, model for B19V | ||||
| n.d. not determined | ||||
|
||||
| Enveloped Viruses | ||||
| HIV | ≥ 5.9 | ≥ 5.9 | ≥ 6.6 | ≥ 18.4 |
| BVDV | ≥ 8.5 | 2.2 | ≥ 6.0 | ≥ 16.7 |
| PRV | 3.8 | 7.2 | ≥ 6.6 | ≥ 17.6 |
| WNV | ≥ 7.4 | n.d. | ≥ 8.1 | ≥ 15.5 |
| Non-Enveloped Viruses | ||||
| HAV | 4.0 | 1.8 | ≥ 6.1 | ≥ 11.9 |
| CPV | [0.5]* | 1.5 | 6.5 | 8.0 |
12. Kcentra - Clinical Pharmacology
12.1 Mechanism of Action
KCENTRA contains the Vitamin K-dependent coagulation Factors II (FII), VII (FVII), IX (FIX), and X (FX), together known as the Prothrombin Complex, and the antithrombotic Protein C and Protein S.
A dose-dependent acquired deficiency of the Vitamin K-dependent coagulation factors occurs during Vitamin K antagonist treatment. Vitamin K antagonists exert anticoagulant effects by blocking carboxylation of glutamic acid residues of the Vitamin K-dependent coagulation factors during hepatic synthesis, lowering both factor synthesis and function. The administration of KCENTRA rapidly increases plasma levels of the Vitamin K-dependent coagulation Factors II, VII, IX, and X as well as the antithrombotic Proteins C and S.
Coagulation Factor II
Factor II (prothrombin) is converted to thrombin by activated FX (FXa) in the presence of Ca2+, FV, and phospholipids.
Coagulation Factor VII
Factor VII (proconvertin) is converted to the activated form (FVIIa) by splitting of an internal peptide link. The FVIIa-TF complex activates Factor IX and initiates the primary coagulation pathway by activating FX in the presence of phospholipids and calcium ions.
Coagulation Factor IX
Factor IX (antihemophilic globulin B, or Christmas factor) is activated by the FVIIa-TF complex and by FXIa. Factor IXa in the presence of FVIIIa activates FX to FXa.
Coagulation Factor X
Factor X (Stuart-Prower factor) activation involves the cleavage of a peptide bond by the FVIIIa-Factor IXa complex or the TF-FVIIa complex. Factor Xa forms a complex with activated FV (FVa) that converts prothrombin to thrombin in the presence of phospholipids and calcium ions.
12.2 Pharmacodynamics
International Normalized Ratio (INR)
In the plasma-controlled RCT in acute major bleeding, the INR was determined at varying time points after the start or end of infusion, depending upon study design. The median INR was above 3.0 prior to the infusion and dropped to a median value of 1.20 by the 30 minute time point after start of KCENTRA infusion. By contrast, the median value for plasma was 2.4 at 30 minutes after the start of infusion. The INR differences between KCENTRA and plasma were statistically significant in randomized plasma-controlled trial in bleeding up to 12 hours after start of infusion [see Table 8].
The relationship between these or other INR values and clinical hemostasis in patients has not been established [see Clinical Studies (14)].
| Study | Treatment | Baseline | 30 min | 1 hr | 2-3 hr | 6-8 hr | 12 hr | 24 hr |
|---|---|---|---|---|---|---|---|---|
| INR = international normalized ratio; NC = not collected. | ||||||||
|
||||||||
| Acute Major Bleeding Study | KCENTRA (N = 98) | 3.90 (1.8–20.0) | 1.20*
(0.9–6.7) | 1.30*
(0.9–5.4) | 1.30*
(0.9–2.5) | 1.30*
(0.9–2.1) | 1.20*
(0.9–2.2) | 1.20 (0.9–3.8) |
| Plasma (N = 104) | 3.60 (1.9–38.9) | 2.4 (1.4–11.4) | 2.1 (1.0–11.4) | 1.7 (1.1–4.1) | 1.5 (1.0–3.0) | 1.4 (1.0–3.0) | 1.3 (1.0–2.9) |
|
| Urgent Surgery/ Invasive Procedures Study | KCENTRA (N = 87) | 2.90 (2.0–17.0) | 1.30*
(0.9–7.0) | 1.20*
(0.9–2.5) | 1.30*
(0.9–39.2) | 1.30*
(1.0–10.3) | NC | 1.20 (0.9–2.7) |
| Plasma (N = 81) | 2.90 (2.0–26.7) | 2.15 (1.4–5.4) | 1.90 (1.3–5.7) | 1.70 (1.1–3.7) | 1.60 (1.0–5.8) | NC | 1.30 (1.0–2.7) |
|
12.3 Pharmacokinetics
Fifteen healthy subjects received 50 units/kg of KCENTRA. No subjects were receiving VKA therapy or were experiencing acute bleeding. A single intravenous KCENTRA infusion produced a rapid and sustained increase in plasma concentration of Factors II, VII, IX and X as well as Proteins C and S. The PK analysis [see Table 9] shows that factor II had the longest half-life (59.7 hours) and factor VII the shortest (4.2 hours) in healthy subjects. PK parameters obtained from data derived from the study of healthy subjects may not be directly applicable to patients with INR elevation due to VKA anticoagulation therapy.
| Parameter | Factor IX | Factor II | Factor VII | Factor X | Protein C | Protein S |
|---|---|---|---|---|---|---|
| Terminal half-life (h) | 42.4 (41.6) | 60.4 (25.5) | 5.0 (1.9) | 31.8 (8.7) | 49.6 (32.7) | 50.4 (13.4) |
| IVR (%/units/kg bw)* | 1.6 (0.4) | 2.2 (0.3) | 2.5 (0.4) | 2.2 (0.4) | 2.9 (0.3) | 2.0 (0.3) |
| AUC (IU/dL × h) | 1850.8 (1001.4) | 7282.2 (2324.9) | 512.9 (250.1) | 6921.5 (1730.5) | 5397.5 (2613.9) | 3651.6 (916.3) |
| Clearance (mL/ kg × h) | 3.7 (1.6) | 1.0 (0.3) | 7.4 (4.1) | 1.3 (0.3) | 1.5 (0.9) | 1.2 (0.3) |
| MRT (h)† | 47.3 (49.5) | 82.0 (34.2) | 7.1 (2.7) | 45.9 (12.6) | 62.4 (42.1) | 70.3 (18.3) |
| Vdss (mL/kg)‡ | 114.3 (54.6) | 71.4 (13.7) | 45.0 (10.7) | 55.5 (6.7) | 62.2 (17.4) | 78.8 (11.6) |
The mean in vivo recovery (IVR) of infused factors was calculated in subjects who received KCENTRA. The IVR is the increase in measurable factor levels in plasma (units/dL) that may be expected following an infusion of factors (units/kg) administered as a dose of KCENTRA. The in vivo recovery ranged from 1.15 (Factor IX) to 2.81 (Protein S) [see Table 10].
| Parameter | Incremental (units/dL per units/kg b.w.) | |||
|---|---|---|---|---|
| Acute Major Bleeding Study (N = 98) | Urgent Surgery/Invasive Procedures Study (N = 87) |
|||
| Mean (SD) | 95% CI† | Mean (SD) | 95% CI† | |
| Factor IX | 1.29 (0.71) | (1.14–1.43) | 1.15 (0.57) | (1.03–1.28) |
| Factor II | 2.00 (0.88) | (1.82–2.18) | 2.14 (0.74) | (1.98–2.31) |
| Factor VII | 2.15 (2.96) | (1.55–2.75) | 1.90 (4.50) | (0.92–2.88) |
| Factor X | 1.96 (0.87) | (1.79–2.14) | 1.94 (0.69) | (1.79–2.09) |
| Protein C | 2.04 (0.96) | (1.85–2.23) | 1.88 (0.68) | (1.73–2.02) |
| Protein S | 2.17 (1.66) | (1.83–2.50) | 2.81 (1.95) | (2.38–3.23) |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals to evaluate the carcinogenic potential of KCENTRA, or studies to determine the effects of KCENTRA on genotoxicity or fertility have not been performed. An assessment of the carcinogenic potential of KCENTRA was completed and suggests minimal carcinogenic risk from product use.
14. Clinical Studies
Acute Major Bleeding RCT
The efficacy of KCENTRA has been evaluated in a prospective, open-label, (blinded assessor), active-controlled, non-inferiority, multicenter RCT in subjects who had been treated with VKA therapy and who required urgent replacement of their Vitamin K-dependent clotting factors to treat acute major bleeding. A total of 216 subjects with acquired coagulation factor deficiency due to oral Vitamin K antagonist therapy were randomized to a single dose of KCENTRA or plasma. Two hundred twelve (212) subjects received KCENTRA or plasma for acute major bleeding in the setting of a baseline INR ≥ 2.0 and recent use of a VKA anticoagulant. The doses of KCENTRA (25 units/kg, 35 units/kg, or 50 units/kg) based on nominal Factor IX content and plasma (10 mL/kg, 12 mL/kg, or 15 mL/kg) were calculated according to the subject's baseline INR (2–< 4, 4–6, > 6, respectively). The observation period lasted for 90 days after the infusion of KCENTRA or plasma. The modified efficacy (ITT-E) population for KCENTRA included 98 subjects and for plasma included 104 subjects. Additionally, intravenous Vitamin K was administered.
The efficacy endpoint was hemostatic efficacy for the time period from the start of infusion of KCENTRA or plasma until 24 hours. Efficacy was adjudicated as "effective" or "not effective" by a blinded, independent Endpoint Adjudication Board for all subjects who received study product. Criteria for effective hemostasis were based upon standard clinical assessments including vital signs, hemoglobin measurements, and CT assessments at pre-defined time points, as relevant to the type of bleeding (i.e., gastrointestinal, intracranial hemorrhage, visible, musculoskeletal, etc.). The proportion of subjects with effective hemostasis was 72.4% in the KCENTRA group and 65.4% in the plasma group. The lower limit of the 95% confidence interval (CI) for the difference in proportions of KCENTRA minus plasma was -5.8%, which exceeded -10% and thereby demonstrated the non-inferiority of KCENTRA versus plasma (the study's primary objective) [see Table 11]. Because the lower limit of the CI was not greater than zero, the prospectively defined criterion for superiority of KCENTRA for hemostatic efficacy (a secondary objective) was not met.
| Rating | No. (%) of subjects [95% CI] | Difference KCENTRA – Plasma (%) [95% CI]* |
|
|---|---|---|---|
| KCENTRA (N = 98) | Plasma (N = 104) |
||
| CI = confidence interval; N = number of subjects | |||
|
|||
| "Effective" hemostasis | 71 (72.4%) [62.3; 82.6] | 68 (65.4%) [54.9; 75.8] | (7.1%) [-5.8; 19.9] |
Results of a post-hoc analysis of hemostatic efficacy stratified by actual dose of KCENTRA or plasma administered in the acute major bleeding RCT are presented in Table 12.
| Low Dose | Mid Dose | High Dose | |
|---|---|---|---|
| N = 49 (K) | N = 22 (K) | N = 26 (K) | |
| N = 55 (P) | N = 18 (P) | N = 31 (P) | |
|
|||
| KCENTRA | 36 (74.5%) | 16 (72.7%) | 18 (69.2%) |
| Plasma | 38 (69.1%) | 11 (61.1%) | 19 (61.3%) |
| Difference* | (4.4%) | (11.6%) | (7.9%) |
| 95% CI K–P | -13.2–21.9 | -17.4–40.6 | -17.0–32.9 |
An additional endpoint was the reduction of INR to ≤ 1.3 at 30 minutes after the end of infusion of KCENTRA or plasma for all subjects that received study product. The proportion of subjects with this decrease in INR was 62.2% in the KCENTRA group and 9.6% in the plasma group. The 95% confidence interval for the difference in proportions of KCENTRA minus plasma was 39.4% to 65.9%. The lower limit of the 95% CI of 39.4% demonstrated superiority of KCENTRA versus plasma for this endpoint [see Table 13].
| Rating | No. (%) of subjects [95% CI] | Difference KCENTRA – Plasma (%) [95% CI]* |
|
|---|---|---|---|
| KCENTRA (N = 98) | Plasma (N = 104) |
||
| CI = confidence interval; INR = international normalized ratio; N = total subjects | |||
|
|||
| Decrease of INR to ≤ 1.3 at 30 min | 61 (62.2%) [52.6; 71.8] | 10 (9.6%) [3.9; 15.3] | (52.6%) [39.4; 65.9] |
Urgent Surgery/Invasive Procedure RCT
The efficacy of KCENTRA has been evaluated in a prospective, open-label, active-controlled, non-inferiority, multicenter RCT in subjects who had been treated with VKA therapy and who required urgent replacement of their Vitamin K-dependent clotting factors because of their need for an urgent surgery/invasive procedure. A total of 181 subjects with acquired coagulation factor deficiency due to oral Vitamin K antagonist therapy were randomized to a single dose of KCENTRA or plasma. One hundred seventy-six (176) subjects received KCENTRA or plasma because of their need for an urgent surgery/invasive procedure in the setting of a baseline INR ≥ 2.0 and recent use of a VKA anticoagulant. The doses of KCENTRA (25 units/kg, 35 units/kg, or 50 units/kg) based on nominal Factor IX content and plasma (10 mL/kg, 12 mL/kg, or 15 mL/kg) were calculated according to the subject's baseline INR (2–< 4, 4–6, > 6, respectively). The observation period lasted for 90 days after the infusion of KCENTRA or plasma. The modified efficacy (ITT-E) population for KCENTRA included 87 subjects and for plasma included 81 subjects. Additionally, oral or intravenous Vitamin K was administered.
The efficacy endpoint was hemostatic efficacy for the time period from the start of infusion of KCENTRA or plasma until the end of the urgent surgery/invasive procedure. Criteria for effective hemostasis were based upon the difference between predicted and actual blood losses, subjective hemostasis rating, and the need for additional blood products containing coagulation factors. The proportion of subjects with effective hemostasis was 89.7% in the KCENTRA group and 75.3% in the plasma group. The lower limit of the 95% confidence interval (CI) for the difference in proportions of KCENTRA minus plasma was 2.8%, which exceeded -10% and thereby demonstrated the non-inferiority of KCENTRA versus plasma (the study's primary objective) [see Table 14]. Because the lower limit of the CI was greater than 0, the prospectively defined criterion for superiority of KCENTRA for hemostatic efficacy (a secondary objective) was also met.
| Rating | No. (%) of subjects [95% CI] | Difference KCENTRA – Plasma (%) [95% CI]* |
|
|---|---|---|---|
| KCENTRA (N = 87) | Plasma (N = 81) |
||
| CI = confidence interval; N = number of subjects | |||
|
|||
| "Effective" hemostasis | 78 (89.7%) [83.3; 96.1] | 61 (75.3%) [65.9; 84.7] | (14.3%) [2.8; 25.8] |
Results of a post-hoc analysis of hemostatic efficacy stratified by actual dose of KCENTRA or plasma administered in the urgent surgery/invasive procedure RCT are presented in Table 15.
| Low Dose | Mid Dose | High Dose | |
|---|---|---|---|
| N = 69 (K) | N = 10 (K) | N = 8 (K) | |
| N = 62 (P) | N = 10 (P) | N = 9 (P) | |
|
|||
| KCENTRA | 63 (91.3%) | 8 (80.0%) | 7 (87.5%) |
| Plasma | 48 (77.4%) | 7 (70.0%) | 6 (66.7%) |
| Difference* | (13.9%) | (10.0%) | (20.8%) |
| 95% CI K–P | 1.4–26.6 | -26.5–43.5 | -19.8–53.7 |
An additional endpoint was the reduction of INR to ≤ 1.3 at 30 minutes after the end of infusion of KCENTRA or plasma for all subjects that received study product. The proportion of subjects with this decrease in INR was 55.2% in the KCENTRA group and 9.9% in the plasma group. The 95% confidence interval for the difference in proportions of KCENTRA minus plasma was 31.9% to 56.4%. The lower limit of the 95% CI of 31.9% demonstrated superiority of KCENTRA versus plasma for this endpoint [see Table 16]. The relationship between a decrease in INR to less than or equal to 1.3 and clinical hemostatic efficacy has not been established.
| Rating | No. (%) of subjects [95% CI] | Difference KCENTRA – Plasma (%) [95% CI]* |
|
|---|---|---|---|
| KCENTRA (N = 87) | Plasma (N = 81) |
||
| CI = confidence interval; INR = international normalized ratio; N = total subjects | |||
|
|||
| Decrease of INR to ≤ 1.3 at 30 min | 48 (55.2%) [44.7; 65.6] | 8 (9.9%) [3.4; 16.4] | (45.3%) [31.9; 56.4] |
The European Bleeding and Surgical Study was an open-label, single-arm, multicenter study.1 Forty-three (43) subjects who were receiving VKA were treated with KCENTRA, because they either (1) required a surgical or an invasive diagnostic intervention (26 subjects), or (2) experienced an acute bleeding event (17 subjects). The dose of KCENTRA (25 units/kg, 35 units/kg, or 50 units/kg) based on nominal Factor IX content was calculated according to the subject's baseline INR value (2–< 4, 4–6, > 6). The endpoint was the decrease of the INR to ≤ 1.3 within 30 minutes after end of KCENTRA infusion in subjects who received any portion of study product.
Of the 17 evaluable subjects receiving KCENTRA for acute bleeding, 16 subjects (94%) experienced a decrease in INR to ≤ 1.3 within 30 minutes after the end of the KCENTRA infusion.
In RCTs, levels of Coagulation Factors II, VII, IX, X, and Antithrombotic Proteins C and S were measured after the infusion of KCENTRA or plasma and the results were similar for subjects with acute major bleeding or subjects requiring an urgent surgery or invasive procedure. In the plasma-controlled RCT in acute major bleeding, the mean duration of KCENTRA infusion was 24 minutes (± 32 minutes) and the mean duration of infusion for plasma was 169 minutes (± 143 minutes). The mean infusion volume of KCENTRA was 105 mL ± 37 mL and the mean infusion volume of plasma was 865 mL ± 269 mL. In the plasma-controlled RCT for patients needing urgent surgery/invasive procedures, the mean duration of KCENTRA infusion was 21 minutes (± 14 minutes) and the mean duration of infusion for plasma was 141 minutes (± 113 minutes). The mean infusion volume of KCENTRA was 90 mL ± 32 mL and the mean infusion volume of plasma was 819 mL ± 231 mL.
The increase in mean factor levels over time following KCENTRA and plasma administration in the plasma-controlled RCT in acute major bleeding is shown in Figure 9 below (the mean factor levels over time following KCENTRA and plasma administration in the plasma-controlled RCT for patients needing urgent surgery/invasive procedures are not shown, but showed similar profiles). Levels of some factors continued to increase at later time points, consistent with the effect of concomitant Vitamin K treatment. Formal pharmacokinetic parameters were not derived because of the effect of Vitamin K on factor levels at time points required for pharmacokinetic profiling.
Figure 9: Mean Factor Levels (Factors II, VII, IX, X, Proteins C & S) over 24 hours in Acute Major Bleeding RCT
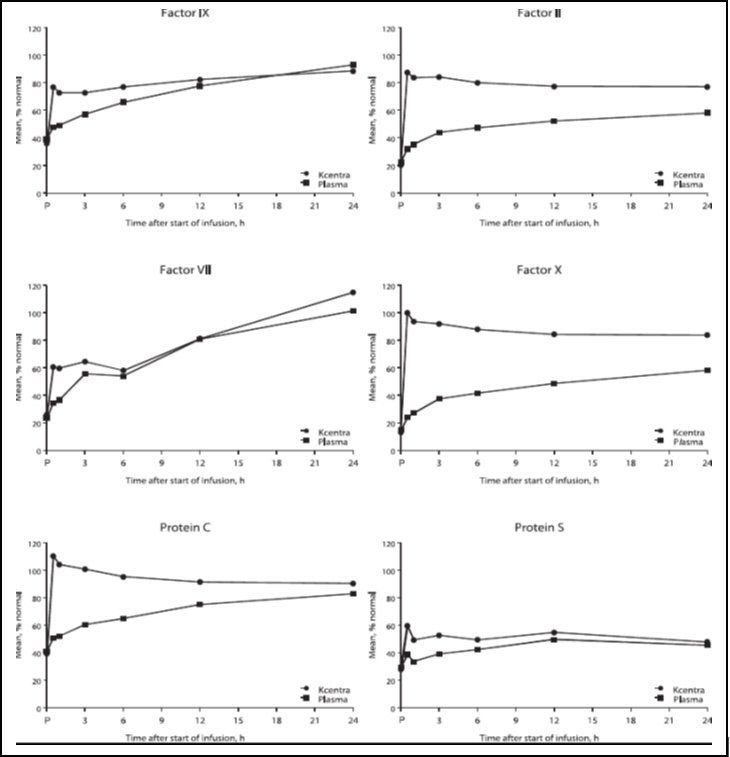
Time axis is scheduled measuring time: hours after start of infusion (P=pre-infusion)
15. References
- Pabinger I, Brenner B, Kalina U, et al. Prothrombin complex concentrate (Beriplex P/N) for emergency anticoagulation reversal: a prospective multinational clinical trial. Journal of Thrombosis and Haemostasis 2008; 6: 622-631.
16. How is Kcentra supplied
- KCENTRA is supplied in a single-dose vial.
- The actual units of potency of all coagulation factors (Factors II, VII, IX and X), Proteins C and S in units are stated on each KCENTRA carton.
- The KCENTRA packaging components are not made with natural rubber latex.
Table 17. How Supplied
Each kit consists of the following:
| Carton NDC Number | Components |
|---|---|
| 63833-386-02 |
|
| 63833-387-02 |
|
Storage and Handling
Prior to Reconstitution
- KCENTRA is for single-dose only. Contains no preservatives.
- Store KCENTRA between 2–25°C (36–77°F), this includes room temperature, not to exceed 25°C (77°F). Do not freeze.
- KCENTRA is stable for 36 months from the date of manufacture, up to the expiration date on the carton and vial labels.
- Do not use KCENTRA beyond the expiration date on the vial label and carton.
- Store the vial in the original carton to protect it from light.
17. Patient Counseling Information
- Inform patients of the signs and symptoms of allergic hypersensitivity reactions, such as urticaria, rash, tightness of the chest, wheezing, hypotension and/or anaphylaxis experienced during or after injection of KCENTRA [see Warnings and Precautions (5.1)].
- Inform patients of signs and symptoms of thrombosis, such as limb or abdomen swelling and/or pain, chest pain or pressure, shortness of breath, loss of sensation or motor power, altered consciousness, vision, or speech [see Warnings and Precautions (5.2)].
- Inform patients that, because KCENTRA is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent, and theoretically, the Creutzfeldt-Jakob disease (CJD) agent [see Warnings and Precautions (5.3) and Description (11)].
Manufactured by:
CSL Behring GmbH
35041 Marburg Germany
US License No. 1765
Distributed by:
CSL Behring LLC
Kankakee, IL 60901 USA
Mix2Vial® is a registered trademark of West Pharma. Services IL, Ltd., a subsidiary of West Pharmaceuticals Services, Inc.
PRINCIPAL DISPLAY PANEL - Kit Carton - 500 U
NDC 63833-386-02
500 U Range
Prothrombin Complex
Concentrate (Human)
Kcentra®
One single-dose vial containing 400 – 620 units of lyophilized Factor IX concentrate for
reconstitution. Also contains Factors II, VII, X and proteins C and S.
For Intravenous Administration Only
Because Kcentra® is made from human blood, it may carry a risk of transmitting
infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent,
and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent.
See package insert Warnings and Precautions section.
CSL Behring

PRINCIPAL DISPLAY PANEL - Kit Carton - 1000 U
NDC 63833-387-02
1000 U Range
Prothrombin Complex
Concentrate (Human)
Kcentra®
One single-dose vial containing 800 – 1240 units
of lyophilized Factor IX concentrate for reconstitution.
Also contains Factors II, VII, X and proteins C and S.
For Intravenous Administration Only
Because Kcentra® is made from human blood, it may carry a risk of
transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob
disease (vCJD) agent, and, theoretically, the Creutzfeldt-Jakob disease
(CJD) agent. See package insert Warnings and Precautions section.
CSL Behring

| KCENTRA
prothrombin, coagulation factor vii human, coagulation factor ix human, coagulation factor x human, protein c, protein s human, and water kit |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| KCENTRA
prothrombin, coagulation factor vii human, coagulation factor ix human, coagulation factor x human, protein c, protein s human, and water kit |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - CSL Behring GmbH (326530474) |
| Registrant - CSL Behring LLC (058268293) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| CSL Behring GmbH | 326530474 | MANUFACTURE | |
More about Kcentra (prothrombin complex)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: anticoagulant reversal agents
- En español