Iqirvo: Package Insert / Prescribing Info
Package insert / product label
Generic name: elafibranor
Dosage form: tablet, film coated
Drug class: Miscellaneous metabolic agents
Medically reviewed by Drugs.com. Last updated on Jun 15, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
IQIRVO (elafibranor) tablets, for oral use
Initial U.S. Approval: 2024
Indications and Usage for Iqirvo
IQIRVO is a peroxisome proliferator-activated receptor (PPAR) agonist indicated for the treatment of primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults who have an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA.
This indication is approved under accelerated approval based on reduction of alkaline phosphatase (ALP). Improvement in survival or prevention of liver decompensation events have not been demonstrated. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s). (1)
Limitations of Use
Use of IQIRVO is not recommended in patients who have or develop decompensated cirrhosis (e.g., ascites, variceal bleeding, hepatic encephalopathy). (8.7, 12.3)
Iqirvo Dosage and Administration
Dosage Forms and Strengths
Tablets: 80 mg (3)
Contraindications
None. (4)
Warnings and Precautions
- Myalgia, Myopathy, and Rhabdomyolysis: Assess for muscle pain and myopathy prior to IQIRVO initiation. Consider periodic assessment (clinical exam, CPK measurement). Interrupt IQIRVO if there is new onset or worsening of muscle injury, or muscle pain. (5.1)
- Fractures: The risk of fracture should be considered in the care of patients treated with IQIRVO. Apply current standards of care for assessing and maintaining bone health. (5.2)
- Adverse Effects on Fetal and Newborn Development: May cause fetal harm. Verify that a female of reproductive potential is not pregnant prior to initiating IQIRVO. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.3, 8.1, 8.3)
- Drug-Induced Liver Injury: Obtain clinical and laboratory assessments at treatment initiation and monitor thereafter according to routine patient management. Interrupt the treatment if liver tests worsen, or patients develop signs and symptoms consistent with clinical hepatitis. Consider permanent discontinuation if liver tests worsen after restarting IQIRVO. (5.4)
- Hypersensitivity Reactions: If severe hypersensitivity reactions occur, permanently discontinue IQIRVO. If a mild or moderate hypersensitivity reaction occurs, interrupt IQIRVO and treat promptly. Monitor until signs and symptoms resolve. (5.5)
- Biliary Obstruction: Avoid use in patients with complete biliary obstruction. If biliary obstruction is suspected, interrupt IQIRVO and treat as clinically indicated. (5.6)
Adverse Reactions/Side Effects
Most common adverse reactions with IQIRVO (reported in ≥ 5% and higher compared to placebo) are weight gain, diarrhea, abdominal pain, nausea, vomiting, arthralgia, constipation, muscle injury, fracture, gastroesophageal reflux disease, dry mouth, weight loss, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Ipsen Biopharmaceuticals, Inc. at 1-855-463-5127 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Hormonal Contraceptives: Switch to effective non-hormonal contraceptives or add a barrier method when using hormonal contraceptives and for at least 3 weeks after last dose. (7.1)
- HMG-CoA Reductase Inhibitors: Monitor for signs and symptoms of muscle injury. (5.1, 7.1)
- Rifampin: Monitor the biochemical response (e.g., ALP and bilirubin) when patients initiate rifampin during IQIRVO treatment. (7.2)
- Bile Acid Sequestrants: Administer at least 4 hours before or 4 hours after taking a bile acid binding sequestrant, or at as great an interval as possible. (2.3, 7.2)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2024
Full Prescribing Information
1. Indications and Usage for Iqirvo
IQIRVO is indicated for the treatment of primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults who have had an inadequate response to UDCA, or as monotherapy in patients unable to tolerate UDCA.
This indication is approved under accelerated approval based on reduction of alkaline phosphatase (ALP) [see Clinical Studies (14)]. Improvement in survival or prevention of liver decompensation events have not been demonstrated. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
Limitations of Use
Use of IQIRVO is not recommended in patients who have or develop decompensated cirrhosis (e.g., ascites, variceal bleeding, hepatic encephalopathy) [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
2. Iqirvo Dosage and Administration
2.1 Recommended Evaluation Before Initiating IQIRVO
Before initiating IQIRVO:
- Evaluate for muscle pain or myopathy [see Warnings and Precautions (5.1)].
- Verify that females of reproductive potential are not pregnant prior to initiating treatment with IQIRVO [see Warnings and Precautions (5.3), Use in Specific Populations (8.1, 8.3)].
2.2 Recommended Dosage and Administration
The recommended dosage of IQIRVO is 80 mg taken orally once daily with or without food [see Clinical Pharmacology (12.3)].
2.3 Administration Modification for Bile Acid Sequestrants
Administer IQIRVO at least 4 hours before or 4 hours after administering the bile acid sequestrant, or at as great an interval as possible [see Drug Interactions (7.2)].
3. Dosage Forms and Strengths
Tablets: 80 mg, round, orange, film-coated tablets, debossed with "ELA 80" on one side and plain on the other side.
5. Warnings and Precautions
5.1 Myalgia, Myopathy, and Rhabdomyolysis
Rhabdomyolysis resulting in acute kidney injury occurred in one IQIRVO-treated patient who had cirrhosis at baseline and was also taking a stable dose of an HMG-CoA reductase inhibitor (statin). Myalgia or myopathy, with or without CPK elevations, occurred in patients treated with IQIRVO alone or treated concomitantly with a stable dose of an HMG-CoA reductase inhibitor [see Adverse Reactions (6.1)].
Assess for myalgia and myopathy prior to IQIRVO initiation. Consider periodic assessment (clinical exam, CPK measurement) during treatment with IQIRVO, especially in those who have signs and symptoms of new onset or worsening of muscle pain or myopathy. Interrupt IQIRVO treatment if there is new onset or worsening of muscle pain, or myopathy, or rhabdomyolysis.
5.2 Fractures
Fractures occurred in 6% of IQIRVO-treated patients compared to no placebo-treated patients [see Adverse Reactions (6.1)].
Consider the risk of fracture in the care of patients treated with IQIRVO and monitor bone health according to current standards of care.
5.3 Adverse Effects on Fetal and Newborn Development
Based on findings from animal reproduction studies, IQIRVO may cause fetal harm when administered during pregnancy. Treatment of pregnant rats with elafibranor at maternal plasma drug exposures lower than or approximately equal to human exposure at the recommended dose resulted in stillbirths, reduced survival, decrease in pup body weight, and/or blue/black discoloration of the caudal section of body.
For females of reproductive potential, verify that the patient is not pregnant prior to initiation of therapy. Advise females of reproductive potential to use effective non-hormonal contraceptives or add a barrier method when using hormonal contraceptives during treatment with IQIRVO and for 3 weeks following the last dose of IQIRVO [see Drug Interactions (7.1), Use in Specific Populations (8.1, 8.3)].
5.4 Drug-Induced Liver Injury
Drug-induced liver injury (DILI) occurred in one patient who took IQIRVO 80 mg once daily [see Adverse Reactions (6.1)] and two patients who took IQIRVO at 1.5-times the recommended dosage. In one patient who developed DILI while taking IQIRVO at 1.5-times the recommended dosage, the clinical presentation was drug-induced autoimmune-like hepatitis (DI-ALH). The median time to onset of elevation in liver tests was 85 days (range: day 57 to 288). In Study 1, increases in transaminases (alanine aminotransferase [ALT] and aspartate aminotransferase [AST] ≥ 5× ULN) occurred in 6% of IQIRVO-treated patients compared to 6% of placebo-treated patients, and total bilirubin (TB) elevation (> 3× ULN) occurred in 2% of IQIRVO-treated patients compared to no placebo-treated patients.
Obtain baseline clinical and laboratory assessments at treatment initiation with IQIRVO and monitor thereafter according to routine patient management. Interrupt IQIRVO treatment if liver tests (ALT, AST, TB, and/or alkaline phosphatase [ALP]) worsen, or the patient develops signs and symptoms consistent with clinical hepatitis (e.g., jaundice, right upper quadrant pain, eosinophilia). Consider permanent discontinuation if liver tests worsen after restarting IQIRVO.
5.5 Hypersensitivity Reactions
Hypersensitivity reactions have occurred in a clinical trial with IQIRVO at 1.5-times the recommended dosage. Three patients (0.2%) had rash or unspecified allergic reaction that occurred 2 to 30 days after IQIRVO initiation, with positive dechallenges and rechallenges. Hypersensitivity reactions resolved after discontinuation of IQIRVO and treatment with steroids and/or antihistamines.
If a severe hypersensitivity reaction occurs, permanently discontinue IQIRVO. If a mild or moderate hypersensitivity reaction occurs, interrupt IQIRVO and treat promptly. Monitor the patient until signs and symptoms resolve. If a hypersensitivity reaction recurs after IQIRVO rechallenge, then permanently discontinue IQIRVO.
5.6 Biliary Obstruction
Avoid use of IQIRVO in patients with complete biliary obstruction. If biliary obstruction is suspected, interrupt IQIRVO and treat as clinically indicated [see Adverse Reactions (6.1)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Myalgia, Myopathy, and Rhabdomyolysis [see Warnings and Precautions (5.1)]
- Fractures [see Warnings and Precautions (5.2)]
- Drug-Induced Liver Injury [see Warnings and Precautions (5.4)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of IQIRVO is based on Study 1 consisting of 161 patients who were randomized to receive IQIRVO 80 mg (n=108) or placebo (n=53) once daily with a median duration of exposure during the double-blind period of 62 weeks (inter quartile range: 52, 84) [see Clinical Studies (14)]. IQIRVO or placebo was administered in combination with UDCA in 95% of patients and as monotherapy in 5% of patients who were unable to tolerate UDCA.
The most common adverse reaction leading to treatment discontinuation was increased CPK (4%).
Common Adverse Reactions
Table 1 presents common adverse reactions that occurred in Study 1.
| Adverse Reaction† | IQIRVO 80 mg Once Daily N = 108 % (n) | Placebo N = 53 % (n) |
|---|---|---|
|
||
| Weight gain‡ | 23% (25) | 21% (11) |
| Diarrhea | 11% (12) | 9% (5) |
| Abdominal pain‡ | 11% (12) | 6% (3) |
| Nausea | 11% (12) | 6% (3) |
| Vomiting | 11% (12) | 2% (1) |
| Arthralgia | 8% (9) | 4% (2) |
| Constipation | 8% (9) | 2% (1) |
| Muscle pain‡ | 7% (8) | 2% (1) |
| Fracture‡ | 6% (7) | 0 |
| Gastroesophageal reflux disease | 6% (7) | 2% (1) |
| Dry mouth | 5% (5) | 2% (1) |
| Weight loss | 5% (5) | 0 |
| Rash‡ | 5% (5) | 4% (2) |
Myalgia, Myopathy, and Rhabdomyolysis
Muscle injury included rhabdomyolysis, CPK elevation with or without myalgia, and myopathy. Rhabdomyolysis and acute kidney injury (AKI) occurred in one IQIRVO-treated patient who had cirrhosis at baseline and was also on a stable dose of an HMG-CoA reductase inhibitor for a year. Median time to development of myalgia was 85.5 days (interquartile range: 29, 291). CPK elevation and/or myalgia occurred in patients on IQIRVO monotherapy as well as in patients who were concomitantly treated with an HMG-CoA reductase inhibitor.
Table 2 presents the frequency of muscle injury related adverse reactions in Study 1.
| Adverse Reaction | IQIRVO 80 mg Once Daily N = 108 % (n) | Placebo N = 53 % (n) |
|---|---|---|
| Creatine phosphokinase (CPK) increased (>3× ULN) | 4% (4)* | 0 |
| Myalgia | 4% (4)* | 2% (1) |
| CPK increased and Myalgia | 1% (1)† | 0 |
| Rhabdomyolysis and AKI‡ | 1% (1)† | 0 |
Less Common Adverse Reactions
Additional adverse reactions that occurred more frequently in the IQIRVO-treated patients compared to placebo, but in less 5% of patients included dizziness, gastroenteritis, increased blood creatinine, and anemia.
Cholelithiasis and Cholecystitis
New onset of cholelithiasis was detected in 3 (3%) IQIRVO-treated patients compared to no placebo-treated patients. The three IQIRVO-treated patients were taking UDCA concomitantly. An additional patient who had gallstones at baseline developed cholecystitis requiring cholecystectomy.
7. Drug Interactions
7.1 Effects of IQIRVO on Other Drugs
Table 3 includes clinically significant drug interactions affecting other drugs.
| Hormonal Contraceptives | |
| Clinical Impact | IQIRVO is a weak CYP3A4 inducer [see Clinical Pharmacology (12.3)]. Co-administration of IQIRVO and hormonal contraceptives (e.g., birth control pills, skin patches, implant) may reduce the systemic exposure of progestin and ethinyl estradiol (CYP3A4 substrates), which may lead to contraceptive failure and/or an increase in breakthrough bleeding. |
| Intervention | Switch to effective non-hormonal contraceptives or add a barrier method when using hormonal contraceptives during treatment with IQIRVO and for at least 3 weeks after last dose [see Warnings and Precautions (5.3), Use in Specific Populations (8.1, 8.3)]. |
| HMG-CoA Reductase Inhibitors | |
| Clinical Impact | CPK elevation and/or myalgia occurred in patients on IQIRVO monotherapy. Co-administration of IQIRVO and HMG-CoA reductase inhibitors (statins) which have a risk of myalgia, can increase the risk of myopathy by a mechanism that has not been fully characterized [see Adverse Reactions (6.1)]. |
| Intervention | Monitor for signs and symptoms of muscle injury. Consider periodic assessment (clinical exam, CPK) during treatment. Interrupt IQIRVO treatment if there is new onset or worsening of muscle pain or myopathy [see Warnings and Precautions (5.1)]. |
7.2 Effects of Other Drugs on IQIRVO
Table 4 includes clinically significant drug interactions affecting IQIRVO.
| Rifampin | |
| Clinical Impact | Co-administration of IQIRVO with rifampin, an inducer of metabolizing enzymes, may reduce the systemic exposure of elafibranor and its active metabolite via increased metabolism and may result in delayed or suboptimal biochemical response [see Clinical Pharmacology (12.3)]. |
| Intervention | Monitor the biochemical response (e.g., ALP and bilirubin) when patients initiate rifampin during treatment with IQIRVO. |
| Bile Acid Binding Sequestrants | |
| Clinical Impact | Bile acid sequestrants may interfere with the action of IQIRVO by reducing its absorption and systemic exposure, which may reduce IQIRVO efficacy. |
| Intervention | Administer IQIRVO at least 4 hours before or 4 hours after taking a bile acid binding sequestrant, or at as great an interval as possible [see Dosage and Administration (2.3)]. |
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on data from animal reproduction studies, IQIRVO may cause fetal harm when administered during pregnancy. Treatment of pregnant rats with elafibranor during organogenesis through lactation resulted in stillbirths, reduced survival, decrease in pup body weight, and/or blue/black discoloration of the caudal section of body, which occurred at maternal plasma drug exposures lower than or approximately equal to human exposure at the recommended dose (see Data). There are insufficient data from human pregnancies exposed to IQIRVO to allow an assessment of a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Report pregnancies to Ipsen Pharmaceuticals, Inc. Adverse Event reporting line at 1-855-463-5127 and [https://www.ipsen.com/contact-us/].
Data
Animal Data
No effects on embryo-fetal development were observed in pregnant rats treated orally with up to 300 mg/kg/day elafibranor (15-times the recommended dose based on combined AUC [area under the plasma concentration-time curve] for elafibranor and GFT1007) during the period of organogenesis.
No adverse effects on embryo-fetal development were observed in pregnant rabbits treated orally with doses up to 100 mg/kg/day elafibranor, which produced systemic exposures (combined AUC for elafibranor and GFT1007) during the period of organogenesis that were less than the human exposure. Administration of 300 mg/kg/day (3.9-times the recommended dose based on combined AUC for elafibranor and GFT1007) produced marked maternal toxicity, embryo-lethality, reduced fetal weight, and a low incidence of fetal malformations. Variations in ossification of distal limb bones occurred at 100 mg/kg/day, which was associated with strong signs of maternal toxicity (e.g., body weight loss).
A pre- and postnatal development study was performed using oral administration of 10, 30, or 100 mg/kg/day elafibranor in female rats during organogenesis through lactation. All doses produced a reduction in pup survival (during postnatal days 1-4 at 100 mg/kg/day and postnatal days 5-21 at 10 mg/kg/day and higher), a decrease in pup body weight (dose-dependent, up to -28% on postnatal day 1), blue/black discoloration of the caudal section of body, and developmental delays based on evaluation of physical landmarks and functional tests. The developmental delays were likely caused by the decrease in body weight. Adverse effects in the offspring occurred at maternal exposures at or above 0.6-times the recommended dose based on combined AUC for elafibranor and GFT1007. Stillbirths were observed in the 30 and 100 mg/kg/day groups (1.3 and 4.9-times the recommended dose, respectively, based on combined AUC for elafibranor and GFT1007). Aortic or iliac arterial thrombosis was found in decedent pups from females treated with 30 or 100 mg/kg/day. The surviving adult offspring showed no effects on learning and memory, reflex development, or reproductive capability.
8.2 Lactation
Risk Summary
There are no data available on the presence of elafibranor or its metabolites in human or animal milk, or on effects of the drug on the breastfed infant or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed infant, advise patients not to breastfeed during treatment with IQIRVO, and for 3 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal data, IQIRVO may cause fetal harm when administered during pregnancy [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of IQIRVO have not been established in pediatric patients.
8.5 Geriatric Use
Of the 108 IQIRVO-treated patients with PBC, 25 (23%) were 65 years of age and older, while 1 (1%) was 75 years of age and older. No overall differences in effectiveness of IQIRVO has been observed in patients 65 years of age and older compared to younger adult patients. In healthy elderly subjects (age range 75-80 years), mean systemic exposure (AUC) of elafibranor and the major active metabolite, GFT1007 was 23% and 52% higher, respectively, than those in healthy young subjects (age range 26 to 42 years).
No dosage adjustment for patients 65 years of age and older is necessary. However, because of limited clinical experience with IQIRVO in patients older than 75 years old, closer monitoring of adverse events in patients older than 75 years is recommended [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
The recommended dosage in patients with mild, moderate, or severe renal impairment is the same as in patients with normal kidney function [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage adjustment is recommended for patients with mild hepatic impairment (Child-Pugh A) [see Clinical Pharmacology (12.3)].
The safety and efficacy of IQIRVO in patients with decompensated cirrhosis have not been established. Use of IQIRVO is not recommended in patients who have or develop decompensated cirrhosis (e.g., ascites, variceal bleeding, hepatic encephalopathy). Monitor patients with cirrhosis for evidence of decompensation. Consider discontinuing IQIRVO if the patient progresses to moderate or severe hepatic impairment (Child-Pugh B or C).
11. Iqirvo Description
Elafibranor and its main active metabolite GFT1007 are peroxisome proliferator-activated receptor (PPAR) agonists. Elafibranor is practically insoluble in aqueous media at pH in the range 1.2 to 6.8. It is very slightly soluble at pH 7.5. It is soluble in dichloromethane, freely soluble in DMSO and sparingly soluble in 2-propanol and ethanol.
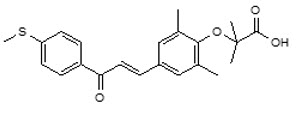
Its chemical formula is C22H24O4S, the molecular weight is 384.49 g/mol, the chemical name is 2-(2,6-Dimethyl-4-{3-[4-(methylsulfanyl)phenyl]-3-oxoprop-1-en-1-yl}phenoxy)-2-methylpropanoic acid and it has the following structural formula:
IQIRVO (elafibranor) tablets are supplied as 80 mg film-coated tablets for oral administration. Each tablet contains 80 mg elafibranor and the following inactive ingredients: colloidal silica dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and povidone. The film coating consists of: iron oxide red, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
12. Iqirvo - Clinical Pharmacology
12.1 Mechanism of Action
Elafibranor and its main active metabolite GFT1007 are peroxisome proliferator-activated receptor (PPAR) agonists, both of which activate PPAR-alpha, PPAR-gamma, and PPAR-delta in vitro. However, the mechanism by which elafibranor exerts its therapeutic effects in patients with PBC is not well understood. Pharmacological activity that is potentially relevant to therapeutic effects includes inhibition of bile acid synthesis through activation of PPAR-alpha and PPAR-delta. The signaling pathway for PPAR-delta was reported to include Fibroblast Growth Factor 21 (FGF21)-dependent downregulation of CYP7A1, the key enzyme for the synthesis of bile acids from cholesterol.
An in vitro PPAR functional assay showed that both elafibranor and GFT1007 produced activation of PPAR-alpha (EC50 = 46 nM and 14 nM, respectively, and Emax = 56% and 61%, respectively, relative to reference agonists). The potency of elafibranor and GFT1007 for PPAR-alpha activation exceeded the respective potencies for PPAR-gamma and PPAR-delta activation by approximately 3- to 8-fold. Although the in vitro pharmacology studies detected PPAR-gamma activation by elafibranor and its metabolite GFT1007, toxicology studies in rats and monkeys (species with plasma metabolite profiles comparable to human) showed none of the adverse effects that are associated with PPAR-gamma activation.
12.2 Pharmacodynamics
In patients with PBC treated with 80 mg once daily of IQIRVO (Study 1), a greater reduction in mean alkaline phosphatase (ALP) from baseline was observed as early as 4 weeks after treatment compared to the placebo group and lower ALP was generally maintained through week 52 [see Clinical Studies (14)].
In another study, there was no apparent dose dependent increase in the reduction of ALP from baseline observed between 80 mg and 120 mg (1.5-times the recommended dose) once daily dosing in patients with PBC.
12.3 Pharmacokinetics
Following once daily dosing, steady state of elafibranor was achieved by Day 14, while steady state of GFT1007 was achieved by Day 7. The pharmacokinetics (PK) of elafibranor and GFT1007 were time-independent after 16-day repeated administration. At steady state, mean AUC0-24h of elafibranor and GFT1007 increased 3.3-fold and 2.6-fold for a 2.5-fold dose increase from 40 mg to 100 mg and 2.9-fold and 2.2-fold, respectively from 120 mg to 300 mg. Mean AUC0-24 of GFT1007 was 3.2-fold higher than the elafibranor exposure in patients with PBC at steady state.
| Cmax,ss (ng/mL) *
Mean (SD) | AUC0-24 (ng ∙ h/mL) *
Mean (SD) | AUC ratio between Day 15/Day 1 Mean (min, max)* |
|
|---|---|---|---|
| Abbreviations: AUC = area under the concentration-versus-time curve; Cmax = maximum concentration; SS= Steady State | |||
|
|||
| Elafibranor | 802 (443) | 3758 (1749) | 2.9 (0.86- to 13) |
| GFT1007 | 2058 (459) | 11985 (7149) | 1.3 (0.6- to 3) |
Absorption
Following once daily dosing of 80 mg in patients with PBC, median time to peak plasma concentrations (Tmax) of elafibranor and GFT1007 was 1.25 hours (range: 0.5-2 hours).
Effect of Food
When administered with a high-fat and high-calorie meal, Tmax was delayed by 30 minutes for elafibranor and by 1-hour for GFT1007 compared to in fasted conditions. Under fed condition, mean Cmax and AUC of elafibranor decreased by 50% and 15% respectively and mean Cmax of GFT1007 decreased by 30%, but the AUC was not affected compared to fasted conditions. The difference was not clinically meaningful.
Distribution
Plasma protein binding of both elafibranor and GFT1007 was approximately 99.7% (mainly to serum albumin). The mean apparent volume of distribution (Vd/F) of elafibranor in healthy subjects was 4731 L, following a single dose of elafibranor at 80 mg in fasted conditions.
Elimination
Following a single 80 mg dose under fasted conditions, median elimination half-life was 70.2 hours (range 37.1 to 92.2 hours) for elafibranor, and 15.4 hours (range 9.39 to 21.7 hours) for major active metabolite GFT1007. Elafibranor mean apparent total clearance (CL/F) was 50.0 L/h after a single 80 mg dose under fasted conditions.
Metabolism
Elafibranor is extensively metabolized to form a major active metabolite, GFT1007. The mean systemic exposure (AUC) to GFT1007 was 3.2-fold higher than that of elafibranor at steady state. Additional major inactive metabolite, an acyl glucuronide conjugate GFT3351 that consisted of four stereoisomers was formed.
In vitro studies showed that elafibranor was metabolized by cytosolic enzyme, 15-ketoprostaglandin 13-Δ reductase (PTGR1), to form GFT1007. Elafibranor was also metabolized by CYP2J2, and uridine diphosphate (UDP)-glucuronosyltransferase (UGT) isoforms, UGT1A3, UGT1A4, and UGT2B7. GFT1007 was further metabolized by CYP2C8 and UGT1A3 and UGT2B7.
Excretion
Following a single 120 mg oral dose (1.5-times the recommended dose) of 14C-radiolabelled elafibranor in healthy subjects, approximately 77.1% of the dose was recovered in feces, primarily as elafibranor (56.7% of the administered dose) and its major metabolite GFT1007 (6.08% of the administered dose). Approximately 19.3% was recovered in urine, primarily as glucuronide conjugate GFT3351 (11.8% of the administered dose). A negligible amount of unchanged elafibranor or GFT1007 was detectable in the urine. Biliary excretion of elafibranor in humans was suggested by the excretion of 60% of orally administered elafibranor in the bile of rats.
Specific Populations
There was no evidence that sex and body mass index (BMI) (14.5 to 53.5 kg/m2) or body weight (43 kg to 120 kg) had any clinically meaningful impact on PK of elafibranor and GFT1007.
Age
Following single dose 120 mg elafibranor administration (1.5-times the recommended dose), the AUCinf of elafibranor and GFT1007 was 23% and 51% higher, respectively in healthy elderly subjects (age range 75-80 years) than in healthy young subjects (age 26-42 years) [see Use in Specific Populations (8.5)].
Patients with Renal Impairment
Following a single dose of 120 mg elafibranor administration (1.5-times the recommended dose), the systemic exposure of elafibranor was 32% lower and GFT1007 was not significantly different between patients with normal renal function and patients with severe renal impairment (eGFR < 15 mL/min/1.73 m2, Modification of Diet in Renal Disease (MDRD)) but not yet on dialysis. The unbound fraction of elafibranor was 21% lower and GFT1007 was not significantly different between patients with normal renal function and patients with severe renal impairment [see Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
Following a single dose 120 mg elafibranor administration (1.5-times the recommended dose), no clinically significant differences in the pharmacokinetics of elafibranor or GFT1007 (mean change < 30%) were observed in patients with hepatic impairment (Child-Pugh A, B and C). However, the unbound fraction of elafibranor and GFT1007 was significantly increased by 2-fold and 2.6-fold, respectively, in patients with severe hepatic impairment (Child-Pugh C) [see Use in Specific Populations (8.7)].
Drug Interactions
Effect of IQIRVO on the Pharmacokinetics of Other Drugs
In vitro Studies:
Elafibranor, GFT1007 and GFT3351 did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. No time dependent CYP inhibition is observed. Elafibranor and GFT1007 did not induce CYP1A2, CYP2B6, and CYP3A4. The CYP induction potential for GFT3351 was not assessed.
Elafibranor is not expected to inhibit UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B7, 2B10, and 2B15 at the clinically relevant concentrations. GFT1007 is not expected to inhibit UGT1A1, 1A3, 1A4, 1A9, 2B7, 2B10, and 2B15 at the clinically relevant concentrations.
GFT1007 inhibited UGT1A6 but the clinical relevance of UGT1A6 inhibition is unknown.
Elafibranor is an inhibitor of bile salt export pump (BSEP) and breast cancer resistance protein (BCRP) and the clinical significance of BSEP and BCRP inhibition by elafibranor is unknown.
GFT3351 is an inhibitor of multidrug resistance associated protein 2 (MRP2) and MRP3 and the clinical significance of MRP2 and MRP3 inhibition by GFT3351 is unknown.
Elafibranor did not inhibit permeability-glycoprotein/multidrug resistance protein 1 (P-gp/MDR1), organic anion transporting polypeptides 1B1 (OATP1B1), organic cation transporter 1 (OCT1), OCT2, organic anion transporter 1 (OAT1), multidrug and toxin extrusion protein 1 (MATE1), MATE2-K and OAT3 and OATP1B3.
GFT1007 did not inhibit OAT3, OATP1B3, BSEP, P-gp/MDR1, BCRP, OATP1B1, OCT1, OCT2, OAT1, MATE1 and MATE2-K. GFT3351 did not inhibit BCRP, P-gp, OATP1B1, OATP1B3, OAT1, OAT2, OAT3, OCT1, OCT2, MATE1, MATE2-K, and BSEP.
Clinical Studies:
Warfarin (CYP2C9 Substrate):
No clinically significant differences in Cmax and AUC of S-warfarin and R-warfarin were observed when a single dose of warfarin 15 mg was administered with elafibranor 120 mg once daily at steady state compared to administered alone. No difference in international normalized ratio (INR) was observed.
Simvastatin (CYP3A4, OATP1B1 and OATP1B3 Substrates):
The active metabolite of simvastatin, simvastatin β-hydroxyacid Cmax decreased by 26% and AUCinf decreased by 32% following concomitant use of a single dose of simvastatin 20 mg and elafibranor 80 mg once daily at steady state. The change in simvastatin β-hydroxyacid exposure was not considered clinically meaningful. The results indicate that IQIRVO is a weak CYP3A4 inducer [see Drug Interaction (7.1)].
Atorvastatin (CYP3A, OATP1B1 and OATP1B3 Substrates):
Atorvastatin Cmax decreased by 28% and AUCinf decreased by 12% following concomitant use of a single dose of atorvastatin 40 mg and elafibranor 180 mg once daily at steady state. The change in atorvastatin exposure was not considered clinically meaningful.
Sitagliptin (dipeptidyl peptidase-IV (DPP-IV) Inhibitor):
In healthy subjects, no significant differences in plasma glucose and glucagon-like peptide-1 (GLP-1) were observed when elafibranor 120 mg was co-administered with sitagliptin 75 mg BID in comparison to administering sitagliptin alone. The relevance of the results to patients is unclear.
Effect of Other Drugs on the Pharmacokinetics of IQIRVO
In vitro Studies:
Elafibranor is a substrate of PTGR1 as well as CYP2J2 and UGT enzymes (e.g., UGT1A3, UGT1A4, and UGT2B7). GFT1007 is a substrate of CYP2C8 and UGT enzymes (e.g., UGT1A3 and UGT2B7).
Elafibranor is a substrate for MRP2 and BCRP. Potential impact of concomitant MRP2 or BCRP inhibitors was not studied in humans; thus, the clinical significance is unknown. GFT1007 is not a substrate for BCRP or MRP2. Neither elafibranor nor GFT1007 is a substrate of P-gp, OATP1B1, OATP1B3, OAT1, OAT3 and OCT2.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 2-year study in CD-1 mice, oral administration of elafibranor produced hepatocellular tumors (adenoma or carcinoma) in both sexes at doses of 1, 3, 10, and 30 mg/kg/day (0.007 to 0.14 times the recommended dose in males and 0.003 to 0.16 times the recommended dose in females based on combined AUC for elafibranor and GFT1007).
In a 2-year study in Sprague-Dawley rats, oral administration of elafibranor produced hepatocellular tumors (adenoma or carcinoma) at 10 mg/kg/day and higher in males (0.36 times the recommended dose based on combined AUC for elafibranor and GFT1007) and at 30 mg/kg/day in females (2.1 times the recommended dose based on combined AUC for elafibranor and GFT1007). In males, elafibranor also produced pancreatic acinar cell adenoma and testicular Leydig cell adenoma at 30 mg/kg/day (2.1 times the recommended dose based on combined AUC for elafibranor and GFT1007).
The liver tumors in mice and rats may be attributed to the expected rodent-specific PPARα-related liver toxicity and its related consequences. Therefore, the relevance to humans is uncertain.
Mutagenesis
Elafibranor was negative in the in vitro bacterial reverse mutation (Ames) assay, the in vivo rat micronucleus assay, and the in vivo rat comet assay. Elafibranor was mutagenic in L5178Y tk+/- mouse lymphoma cells in the absence or presence of metabolic activation and it induced the formation of micronuclei in this cell line in the presence of metabolic activation.
The metabolites GFT1007 and racemic GFT3351 were both negative in the in vitro bacterial reverse mutation (Ames) assay. GFT1007 tested negative in the in vitro micronucleus assay in L5178Y tk+/- mouse lymphoma cells, and GFT3351 tested negative in the in vitro micronucleus assay in human lymphocytes.
The overall data and weight-of-evidence from the comprehensive battery of in vivo and in vitro genotoxicity assays conducted for elafibranor, its principal active metabolite GFT1007, and the acyl glucuronide metabolite racemic GFT3351 indicate that the parent drug and its tested metabolites are unlikely to have genotoxic potential.
14. Clinical Studies
The efficacy of IQIRVO was evaluated in Study 1 (NCT04526665), a multi-center, randomized, double-blind, placebo-controlled study. The study included 161 adults with PBC with an inadequate response or intolerance to UDCA. Patients were randomized to receive IQIRVO 80 mg (n=108) or placebo (n=53) once daily for at least 52 weeks. When applicable, patients continued their pre-study dose of UDCA throughout the study. Patients were included in the study if their ALP was greater than or equal to 1.67-times the ULN and TB was less than or equal to 2-times the ULN. Patients were excluded if they had other liver disease or in case of decompensated cirrhosis.
The mean age of patients in Study 1 was 57 (Range: 36, 76) years, and the mean weight was 70.8 (Range: 43, 134) kg. The study population was predominately female (96%) and White (91%). The baseline mean ALP concentration was 321.9 (Range: 151, 1398) U/L, and 39% of patients had a baseline ALP concentration greater than 3-times the ULN.
The mean baseline TB concentration was 0.56 (Range: 0.15, 1.76) mg/dL, and 96% of patients had a baseline TB concentration less than or equal to ULN. The baseline mean concentration of ALT was 50 (Range: 11 to 188) U/L and mean baseline concentration for AST was 46 (Range: 14 to 203) U/L.
Most patients (95%) received study treatment (IQIRVO or placebo) in combination with UDCA. There were 6 (6%) in the IQIRVO-treated patients and 2 (4%) in the placebo-treated patients who were unable to tolerate UDCA and received IQIRVO as monotherapy. At baseline, 12 (11%) of the IQIRVO-treated patients and 8 (15%) of the placebo-treated patients met at least one of the following criteria: serum albumin < 3.5g/dL, INR >1.3, TB > 1-time ULN, Fibroscan >16.9 kPa, or historical biopsy suggestive of cirrhosis.
The primary endpoint was biochemical response at Week 52, where biochemical response was defined as achieving ALP less than 1.67-times ULN, TB less than or equal to ULN, and ALP decrease greater than or equal to 15% from baseline. The ULN for ALP was defined as 129 U/L for males and 104 U/L for females. The ULN for TB was defined as 1.20 mg/dL. ALP normalization (i.e., ALP less than or equal to ULN) at Week 52 was a key secondary endpoint.
Table 6 presents results at Week 52 for the percentage of patients who achieved biochemical response, achieved each component of biochemical response, and achieved ALP normalization. IQIRVO demonstrated greater improvement on biochemical response and ALP normalization at Week 52 compared to placebo. Overall, 96% of patients had a baseline TB concentration less than or equal to ULN. Therefore, improvement in ALP was the main contributor to the biochemical response rate results at Week 52.
| IQIRVO 80mg Once Daily (N=108) | Placebo (N=53) | Treatment Difference, % (95% CI)† |
|
|---|---|---|---|
|
|||
| Biochemical response rate, n (%)‡ | 55 (51) | 2 (4) | 47 (32, 57) |
| Components of biochemical response | |||
| ALP less than 1.67-times ULN, n (%) | 56 (52) | 5 (9) | 42 (27, 53) |
| Decrease in ALP of at least 15%, n (%) | 81 (75) | 9 (17) | 58 (43, 69) |
| TB less than or equal to ULN, n (%) § | 92 (85) | 44 (83) | 2 (-9, 16) |
| ALP normalization, n (%)¶ | 16 (15) | 0 (0) | 15 (6, 23) |
Figure 1 depicts the mean (95% confidence interval) ALP levels over 52 weeks. There was a trend of lower ALP in the IQIRVO group compared to the placebo group starting by Week 4 through Week 52.
Figure 1: Mean ALP (+/- 95% Confidence Interval) in Adult Patients with PBC Over 52 Weeks in Study 1
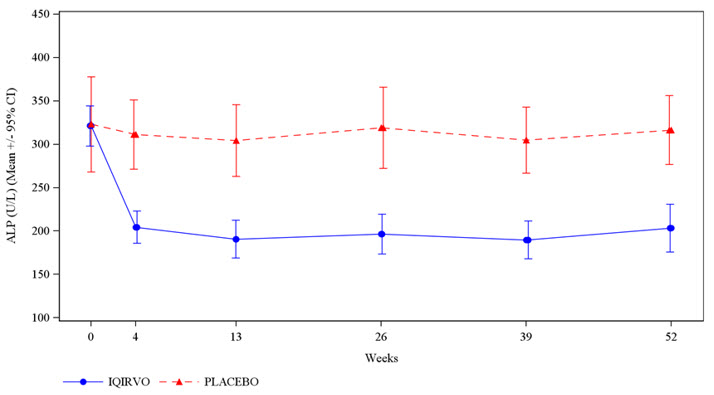
Missing data and data following study treatment discontinuation was imputed by multiple imputation.
16. How is Iqirvo supplied
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Myalgia, Myopathy, and Rhabdomyolysis
Advise patients that IQIRVO may cause rhabdomyolysis. Inform patients to report immediately to their healthcare provider any unexplained muscle symptoms such as pain, soreness, or weakness [see Warnings and Precautions (5.1)].
Fractures
Inform patients or their caregiver(s) that IQIRVO may increase the risk of bone fractures. Advise patients to call their healthcare provider to report any fractures [see Warnings and Precautions (5.2)].
Adverse Effects on Fetal and Newborn Development
Advise pregnant women and females of reproductive potential of the potential risk to the fetus and to inform their healthcare providers of a known, suspected, or planned pregnancy during treatment with IQIRVO. Inform patients to report their pregnancy to Ipsen Biopharmaceuticals, Inc. at (1-855-463-5217). Advise females of reproductive potential to use effective contraception during treatment and for 3 weeks after the last dose of IQIRVO [see Warning and Precautions (5.3), Use in Specific Population (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with IQIRVO and for 3 weeks after the last dose [see Use in Specific Populations (8.2)].
Drug-Induced Liver Injury
Inform patients of the risk of IQIRVO-induced liver injury. Instruct patients to report any signs or symptoms of liver injury (e.g., loss of appetite, nausea, increased fatigue, lower extremity edema, abdominal swelling, or jaundice/icterus) to their healthcare provider [see Warnings and Precautions (5.4)].
Hypersensitivity Reactions
Advise patients to contact their healthcare provider or go to the emergency department if hypersensitivity reactions, such as rash, occur [see Warnings and Precautions (5.5)].
Biliary Obstruction
Instruct patients to immediately report any signs or symptoms of biliary obstruction (e.g., right upper quadrant pain, jaundice) to their healthcare provider so that IQIRVO treatment can be interrupted while the patient is being evaluated [see Warnings and Precautions (5.6)].
Manufactured for: Ipsen Biopharmaceuticals, Inc., One Main Street, 7th Floor, Cambridge, MA, 02142, USA
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Approved: 06/2024 | ||||
| MEDICATION GUIDE
IQIRVO® (eye-ker-vo) (elafibranor) tablets |
|||||
| What is IQIRVO?
IQIRVO is a prescription medicine used to treat primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults who have not responded well to UDCA, or used alone in patients unable to tolerate UDCA. IQIRVO is not recommended for use in people who have symptoms or signs of advanced liver disease (decompensated cirrhosis) including confusion; having fluid in the stomach-area (abdomen); black, tarry, or bloody stools; coughing up or vomiting blood, or having vomit that looks like "coffee grounds." It is not known if taking IQIRVO will improve your chance of survival or prevent liver decompensation. It is not known if IQIRVO is safe and effective in children under 18 years of age. |
|||||
Before taking IQIRVO, tell your healthcare provider about all of your medical conditions, including if you:
Females who can become pregnant:
|
|||||
How should I take IQIRVO?
|
|||||
| What are the possible side effects of IQIRVO? IQIRVO can cause serious side effects including:
|
|||||
|
|
||||
|
|||||
|
|
||||
| Tell your healthcare provider right away if you have any of the following symptoms during treatment with IQIRVO and they are severe or do not go away: | |||||
|
|
||||
|
|||||
|
|
||||
| If you have any of these symptoms stop taking IQIRVO and call your healthcare provider right away or go to the nearest hospital emergency room. | |||||
|
|||||
| The most common side effects of IQIRVO include: | |||||
|
|
|
|||
| Your healthcare provider may tell you to stop taking IQIRVO temporarily or permanently if there are changes to either your liver tests or the level of an enzyme in your blood related to muscle activity called creatine phosphokinase (CPK). Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||||
How should I store IQIRVO?
|
|||||
| General information about the safe and effective use of IQIRVO.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use IQIRVO for a condition for which it was not prescribed. Do not give IQIRVO to other people even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about IQIRVO that is written for health professionals. |
|||||
| What are the ingredients in IQIRVO? Active ingredient: Elafibranor Inactive ingredients: colloidal silica dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and povidone. The film coating consists of: iron oxide red, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. Manufactured for: Ipsen Biopharmaceuticals, Inc., One Main Street, 7th Floor, Cambridge, MA, 02142, USA For more information, 1-855-463-5127 IQIRVO is a registered trademark of Genfit SA © 2024 Ipsen Biopharmaceuticals, Inc. All rights reserved |
|||||
| IQIRVO
elafibranor tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Labeler - Ipsen Biopharmaceuticals, Inc. (118461578) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| PCAS | 276311742 | API MANUFACTURE(15054-0080) , ANALYSIS(15054-0080) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Lab-Service SAS | 269257291 | PARTICLE SIZE REDUCTION(15054-0080) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Quinta-Analytica s.r.o. | 495722555 | ANALYSIS(15054-0080) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| PCAS Finland Oy | 369587311 | ANALYSIS(15054-0080) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Delpharm Milano Srl | 437999026 | MANUFACTURE(15054-0080) , ANALYSIS(15054-0080) , PACK(15054-0080) | |
More about Iqirvo (elafibranor)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: miscellaneous metabolic agents
- En español