Hyqvia: Package Insert / Prescribing Info
Package insert / product label
Generic name: immune globulin 10 percent (human) with recombinant human hyaluronidase
Dosage form: subcutaneous injection
Drug class: Immune globulins
Medically reviewed by Drugs.com. Last updated on Jan 8, 2025.
PRINCIPAL DISPLAY PANEL - 2.5 g/25 mL Vial Label
Immune Globulin Infusion 10% (Human)
Component of HYQVIA
Single-Use Vial
Rx Only
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
U.S. License No. 1898
Infuse 2nd
2.5 g/
25 mL
IG
LE-07-54397
Takeda

PRINCIPAL DISPLAY PANEL - 1.25 mL Vial Label
Recombinant Human Hyaluronidase
Component of HYQVIA
Single-Use Vial
Rx Only
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
Infuse 1st
HY
1.25 mL
160 units per mL
Takeda

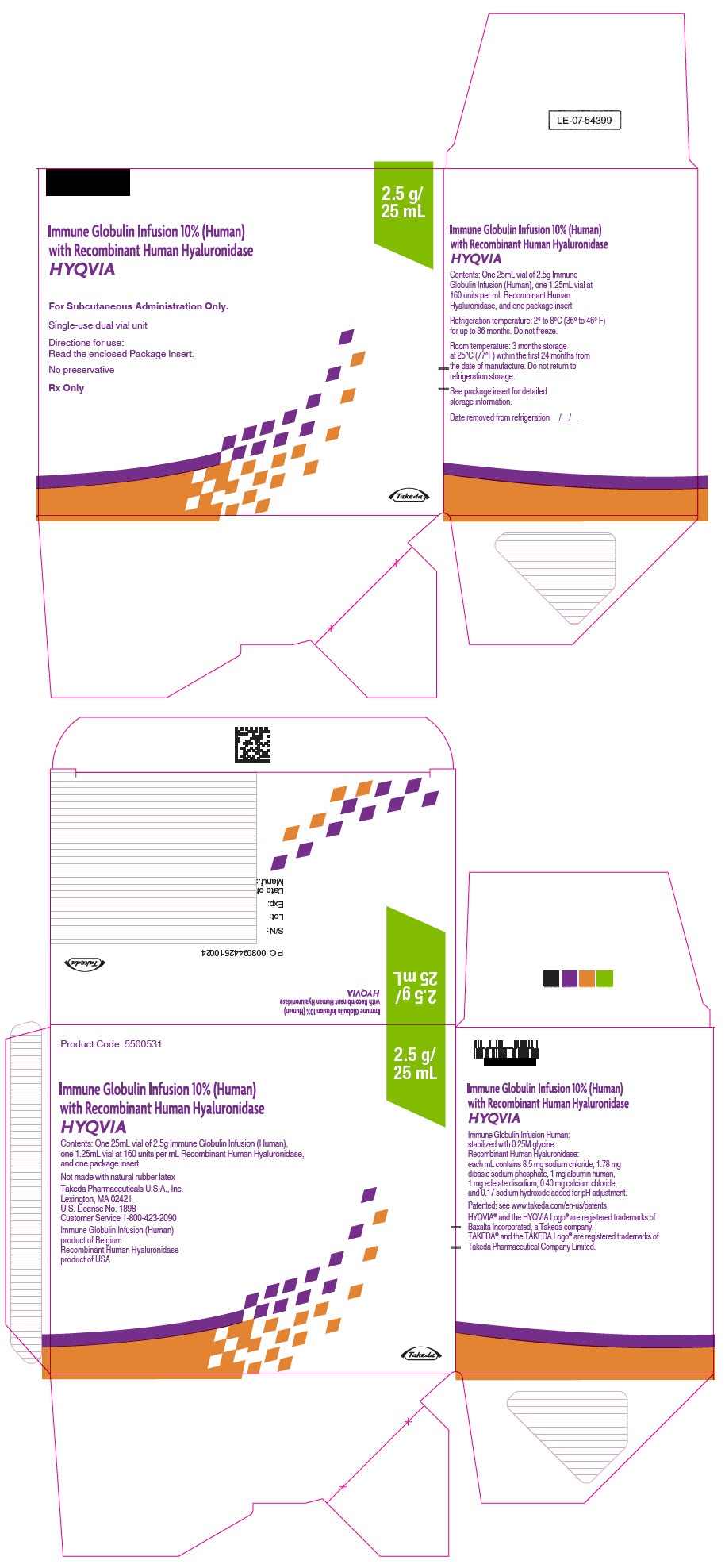
PRINCIPAL DISPLAY PANEL - 2.5 g/25 mL Kit Carton
Immune Globulin Infusion 10% (Human)
with Recombinant Human Hyaluronidase
HYQVIA
For Subcutaneous Administration Only.
Single-use dual vial unit
Directions for use:
Read the enclosed Package Insert.
No preservative
Rx Only
2.5 g/
25 mL
Takeda
PRINCIPAL DISPLAY PANEL - 5 g/50 mL Vial Label
Immune Globulin Infusion 10% (Human)
Component of HYQVIA
Single-Use Vial
Rx Only
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
U.S. License No. 1898
Infuse 2nd
5 g/
50 mL
IG
LE-07-54409
Takeda

PRINCIPAL DISPLAY PANEL - 2.5 mL Vial Label
Recombinant Human Hyaluronidase
Component of HYQVIA
Single-Use Vial
Rx Only
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
Infuse 1st
HY
2.5 mL
160 units per mL
Takeda

PRINCIPAL DISPLAY PANEL - 5 g/50 mL Kit Carton
Immune Globulin Infusion 10% (Human)
with Recombinant Human Hyaluronidase
HYQVIA
For Subcutaneous Administration Only.
Single-use dual vial unit
Directions for use:
Read the enclosed Package Insert.
No preservative
Rx Only
5 g/
50 mL
Takeda

PRINCIPAL DISPLAY PANEL - 10 g/100 mL Vial Label
Immune Globulin Infusion 10% (Human)
Component of HYQVIA
Single-Use Vial
Rx Only
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
U.S. License No. 1898
Infuse 2nd
10 g/
100 mL
IG
LE-07-54412
Takeda

PRINCIPAL DISPLAY PANEL - 5 mL Vial Label
Recombinant Human Hyaluronidase
Component of HYQVIA
Single-Use Vial
Rx Only
Takeda Pharmaceuticals U.S.A., Inc.
Lexington, MA 02421
Infuse 1st
5 mL
160 units per mL
HY
Takeda

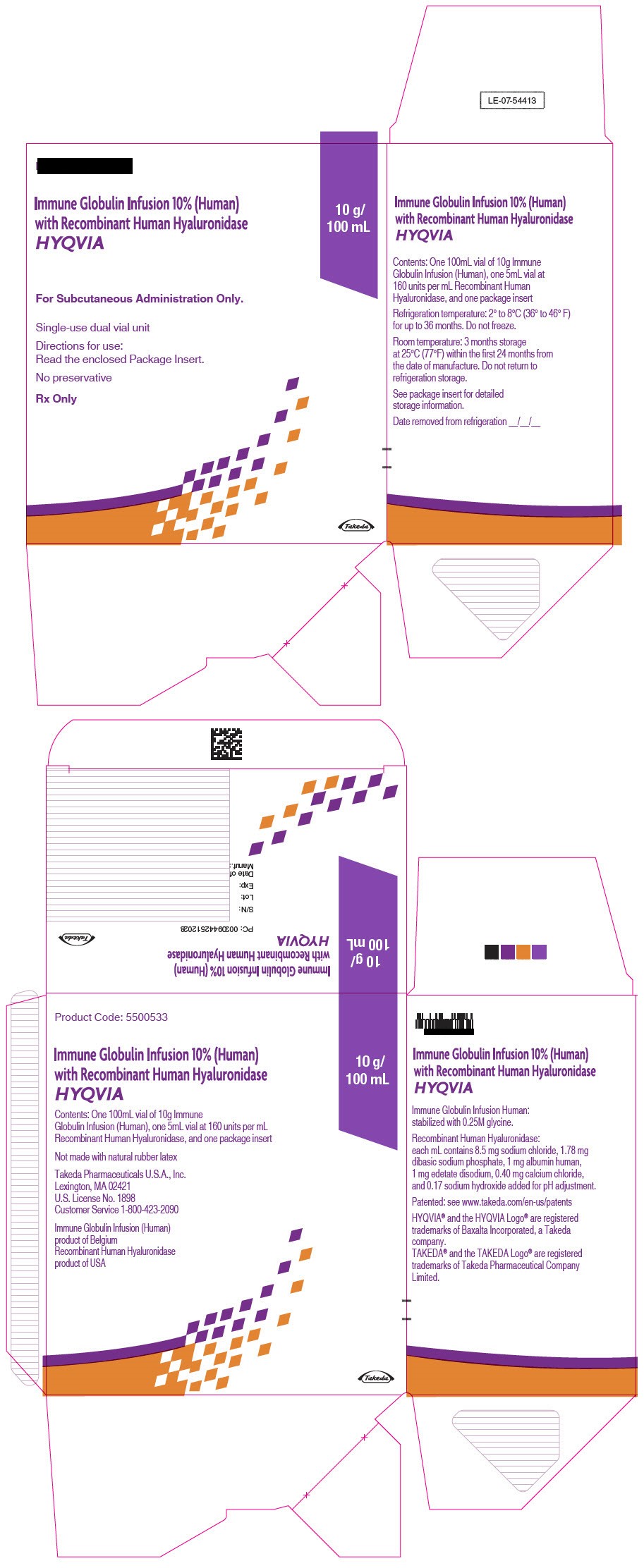
PRINCIPAL DISPLAY PANEL - 10 g/100 mL Kit Carton
Immune Globulin Infusion 10% (Human)
with Recombinant Human Hyaluronidase
HYQVIA
For Subcutaneous Administration Only.
Single-use dual vial unit
Directions for use:
Read the enclosed Package Insert.
No preservative
Rx Only
10 g/
100 mL
Takeda


| HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kit |
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
| HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kit |
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
| HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kit |
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
| HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kit |
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
| HYQVIA
immune globulin infusion 10% (human) with recombinant human hyaluronidase kit |
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
|
||||||||||||||||
| Labeler - Bamboo US BidCo LLC (119087615) |
More about Hyqvia (hyaluronidase / immune globulin)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: immune globulins
- En español