Cuvrior: Package Insert / Prescribing Info
Package insert / product label
Generic name: trientine tetrahydrochloride
Dosage form: tablet, film coated
Drug class: Chelating agents
Medically reviewed by Drugs.com. Last updated on Jun 25, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
CUVRIOR® (trientine tetrahydrochloride) tablets, for oral use
Initial U.S. Approval: 1985
Indications and Usage for Cuvrior
CUVRIOR is a copper chelator indicated for the treatment of adult patients with stable Wilson's disease who are de-coppered and tolerant to penicillamine. (1)
Cuvrior Dosage and Administration
Recommended Dosage and Administration
- Starting total daily dosage of CUVRIOR in adults is 300 mg up to 3,000 mg orally in divided doses (2 times daily). See full prescribing information for recommended conversion table when switching from penicillamine to CUVRIOR. (2.1)
- Total daily dosage of CUVRIOR should not exceed 3,000 mg. (2.1)
- If the number of CUVRIOR tablets prescribed per day cannot be equally divided among doses, then divide total daily dosage such that the higher number of tablets is taken with the first daily dose. (2.1)
- Take CUVRIOR on an empty stomach. (2.2)
- Swallow tablets without crushing, chewing, or dissolving tablets. (2.2)
Switching from Other Trientine Products
- CUVRIOR is not substitutable on a milligram-per-milligram basis with other trientine products. (2.3)
- See full prescribing information for additional information on switching from other trientine products. (2.3)
Clinical Monitoring and Laboratory Monitoring of Copper
Dosage Forms and Strengths
Tablets: 300 mg of trientine tetrahydrochloride, functionally scored. (3)
Contraindications
Hypersensitivity to trientine or to any of the excipients in CUVRIOR. (4)
Warnings and Precautions
- Potential for Worsening of Clinical Symptoms at Initiation of Therapy: May include neurological deterioration. Adjust dosage or discontinue CUVRIOR if clinical condition worsens. (5.1)
- Copper Deficiency: Periodic monitoring is required. (5.2)
- Iron Deficiency: If iron deficiency develops, a short course of iron supplementation may be given. (5.3, 7.1)
- Hypersensitivity Reactions: If rash or other hypersensitivity reaction occurs, consider discontinuing CUVRIOR. (5.4)
Adverse Reactions/Side Effects
Most common adverse reactions (>5%) are abdominal pain, change of bowel habits, rash, alopecia, and mood swings. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Orphalan at 1-800-961-8320 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
-
Mineral Supplements (e.g. iron, zinc, calcium, magnesium): Avoid concomitant use. If concomitant use is unavoidable (2.2, 7.1):
- Iron: Take CUVRIOR at least 2 hours before or 2 hours after iron.
- Other Mineral Supplements: Take CUVRIOR at least 1 hour before or 2 hours after other mineral supplements.
- Other Drugs for Oral Administration: Take CUVRIOR at least 1 hour apart from any other oral drug. (2.2, 7.1)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2024
Full Prescribing Information
1. Indications and Usage for Cuvrior
CUVRIOR is indicated for the treatment of adult patients with stable Wilson's disease who are de-coppered and tolerant to penicillamine.
2. Cuvrior Dosage and Administration
2.1 Recommended Dosage and Administration
The recommended starting total daily dosage of CUVRIOR in adult patients is 300 mg up to 3,000 mg taken orally in divided doses (two times daily). Table 1 provides the recommended starting total daily dosage of CUVRIOR in adult patients switching from penicillamine to CUVRIOR [see Clinical Studies (14)]. Discontinue penicillamine before starting CUVRIOR.
| Penicillamine Total Daily Dosage | CUVRIOR Starting Total Daily Dosage |
|---|---|
| 125 mg | 300 mg |
| 250 mg | 600 mg |
| 375 mg | 900 mg |
| 500 mg | 900 mg |
| 625 mg | 1,200 mg |
| 750 mg | 1,500 mg |
| 875 mg | 1,800 mg |
| 1,000 mg | 2,100 mg |
| 1,125 mg | 2,400 mg |
| 1,250 mg | 2,400 mg |
| 1,375 mg | 2,700 mg |
| 1,500 mg or greater | 3,000 mg |
Adjust the total daily dosage of CUVRIOR according to clinical assessment and laboratory monitoring of copper [see Dosage and Administration (2.4)]. The total daily dosage of CUVRIOR should not exceed 3,000 mg.
If the number of CUVRIOR tablets prescribed per day cannot be equally divided among doses, then divide the total daily dosage such that the higher number of tablets is administered with the first daily dose. Table 2 provides the recommended approach to administration of CUVRIOR tablets to achieve the total daily dosage.
| CUVRIOR | Number of CUVRIOR Tablets to Administer | |
|---|---|---|
| Total Daily Dosage | Morning | Evening |
| 300 mg | 1 | 0 |
| 600 mg | 1 | 1 |
| 900 mg | 2 | 1 |
| 1,200 mg | 2 | 2 |
| 1,500 mg | 3 | 2 |
| 1,800 mg | 3 | 3 |
| 2,100 mg | 4 | 3 |
| 2,400 mg | 4 | 4 |
| 2,700 mg | 5 | 4 |
| 3,000 mg | 5 | 5 |
2.2 Important Administration Instructions
- Discontinue penicillamine before starting CUVRIOR [see Dosage and Administration (2.1)].
- Administer CUVRIOR on an empty stomach, at least 1 hour before meals or 2 hours after meals and at least 1 hour apart from any other food or milk.
- Avoid concomitant use of mineral supplements (e.g. iron, zinc, calcium, magnesium). If concomitant use of mineral supplements is unavoidable [see Drug Interactions (7.1)]:
- Iron supplements: Administer CUVRIOR at least 2 hours before or 2 hours after administration of an iron supplement.
- Other mineral supplements: Administer CUVRIOR at least 1 hour before or 2 hours after administration of other mineral supplements.
- Administer CUVRIOR at least 1 hour apart from any other oral drug.
- Do not remove tablets from the blister pack until just before dosing.
- Swallow tablets of CUVRIOR without crushing, chewing, or dissolving tablets. For patients who have difficulty swallowing the tablet whole, the scored tablet can be divided into two equal halves. Do not store the tablet for future use after the blister has been opened.
- Avoid the use of CUVRIOR in patients who are unable to swallow tablets.
2.3 Switching to CUVRIOR from Other Trientine Products
CUVRIOR is not substitutable on a milligram-per-milligram basis with other trientine products.
If switching a patient from a trientine hydrochloride formulation to CUVRIOR, note that the content of the active moiety (trientine base) is not the same as CUVRIOR. A 250 mg capsule of trientine hydrochloride contains 167 mg of trientine base; in contrast, each 300 mg tablet of CUVRIOR contains 150 mg of trientine base [see Clinical Pharmacology (12.3)].
2.4 Clinical Monitoring and Laboratory Monitoring of Copper
Adjust the total daily dosage of CUVRIOR according to clinical assessment and serum non-ceruloplasmin copper (NCC) levels. Evaluate serum NCC levels when initiating CUVRIOR treatment, after 3 months of treatment and approximately every 6 months thereafter. Therapy may also be monitored periodically (every 6 to 12 months) with measurement of 24-hour urinary copper excretion (UCE) [see Warnings and Precautions (5.1, 5.2)].
3. Dosage Forms and Strengths
Tablets: 300 mg of trientine tetrahydrochloride (equivalent to 150 mg of trientine), oblong, yellow coated, functionally scored, printed with OL75 on each side of score line in black ink. Each large carton contains nine small cartons, each containing a blister pack of 8 tablets (a total of 72 tablets in the large carton).
4. Contraindications
CUVRIOR is contraindicated in patients with hypersensitivity to trientine or to any of the excipients in CUVRIOR [see Warnings and Precautions (5.4)].
5. Warnings and Precautions
5.1 Potential for Worsening of Clinical Symptoms at Initiation of Therapy
Worsening of clinical symptoms, including neurological deterioration, may occur at the beginning of CUVRIOR therapy due to mobilization of excess stores of copper. Adjust the dosage or discontinue CUVRIOR if the patient's clinical condition worsens.
Evaluate serum non-ceruloplasmin copper (NCC) levels when initiating CUVRIOR treatment, after 3 months of treatment and approximately every 6 months thereafter. Therapy may also be monitored periodically (every 6 to 12 months) with measurement of 24-hour urinary copper excretion (UCE) [see Dosage and Administration (2.4)].
5.2 Copper Deficiency
Copper deficiency may develop following treatment with CUVRIOR. Close monitoring for manifestations of copper deficiency is required particularly when copper requirements may change, such as in pregnancy, where appropriate control of copper levels are required to ensure proper growth and mental development [see Dosage and Administration (2.4) and Use in Specific Populations (8.1)].
5.3 Iron Deficiency
Iron deficiency may develop following treatment with CUVRIOR, especially in menstruating or pregnant women, or as a result of the low copper diet recommended for Wilson's disease. If necessary, iron may be given in short courses, but at least two hours should elapse between administration of CUVRIOR and iron [see Drug Interactions (7.1) and Use in Specific Populations (8.1)].
5.4 Hypersensitivity Reactions
Hypersensitivity reactions, characterized by rash, have been reported with the use of trientine. In Trial 1, rash was reported in 12% (3/26) of CUVRIOR-treated patients, and one of these patients discontinued CUVRIOR because of the rash [see Adverse Reactions (6.1)]. If a patient develops a rash or other hypersensitivity reaction during treatment with CUVRIOR, assess clinically and consider discontinuing CUVRIOR [see Contraindications (4)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Potential for Worsening of Clinical Symptoms at Initiation of Therapy [see Warnings and Precautions (5.1)]
- Copper Deficiency [see Warnings and Precautions (5.2)]
- Iron Deficiency [see Warnings and Precautions (5.3)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Common Adverse Reactions
Table 3 presents common adverse reactions over a 24-week period from Trial 1, a prospective, randomized, multi-center study that was conducted in adult patients with Wilson's disease who were de-coppered and tolerant to penicillamine [see Clinical Studies (14)]. Patients were either switched to receive CUVRIOR (N=26) or continued to receive penicillamine (N=27).
Other Adverse Reactions
In Trial 1, anemia developed in 4% (1/26) of CUVRIOR-treated patients and in no patients who continued to receive penicillamine.
In addition, the following adverse reactions have been reported in clinical studies of patients with Wilson's disease who were on therapy with trientine hydrochloride:
- Metabolism and Nutrition Disorders: Iron deficiency
- Musculoskeletal and Connective Tissue Disorders: Systemic lupus erythematosus
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of trientine hydrochloride. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- Gastrointestinal Disorders: Colitis
- Musculoskeletal and Connective Tissue Disorders: Muscle spasms, Rhabdomyolysis
- Nervous System Disorders: Dystonia, Myasthenia gravis
Related/similar drugs
7. Drug Interactions
7.1 Mineral Supplements and Other Oral Drugs
CUVRIOR has the potential to chelate non-copper cations in mineral supplements and other oral drugs, and could be rendered ineffective prior to systemic absorption.
Mineral Supplements
Avoid concomitant use of mineral supplements such as iron, zinc, calcium, or magnesium with CUVRIOR because they may reduce the absorption of CUVRIOR.
However, if iron deficiency develops [see Warnings and Precautions (5.3)], iron supplementation may be given in short courses, but because iron and CUVRIOR each inhibit absorption of the other, administer CUVRIOR at least 2 hours before or 2 hours after administration of an iron supplement [see Dosage and Administration (2.2)].
If concomitant use of other mineral supplements is unavoidable, administer CUVRIOR at least 1 hour before or 2 hours after administration of other mineral supplements.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Available data from published literature and postmarketing experience over several decades with use of trientine for the treatment of Wilson's disease have not identified any drug-associated risks for major birth defects, miscarriages, or other adverse maternal or fetal outcomes. Untreated Wilson's disease may result in worsening disease symptoms during pregnancy and increase the risk of miscarriages in some symptomatic patients (see Clinical Considerations).
In animal reproduction studies, oral administration of trientine in rats during organogenesis resulted in increased embryo-fetal loss at a dose lower than the maximum recommended dose and produced fetal abnormalities at 2.7 times the maximum recommended dose. Copper supplementation in pregnant rats produced a marked reduction in trientine-induced fetal abnormalities. Oral administration of trientine dihydrochloride to pregnant mice during organogenesis increased the percentage of mice with total embryo-fetal loss at approximately 4.3 times the maximum recommended dose and produced fetal abnormalities at approximately 1.1 times the maximum recommended dose. The mechanism of embryo-fetal harm (e.g., copper depletion) was not determined in the mouse study [see Warnings and Precautions (5.2) and Data].
Monitor copper levels throughout pregnancy and use the minimum effective dosage of CUVRIOR throughout pregnancy.
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Untreated Wilson's disease or discontinuation of treatment during pregnancy may result in worsening neurological and hepatic symptoms, including rare reports of hepatic decompensation and liver failure. Untreated Wilson's disease may also increase the risk of miscarriage in some symptomatic patients. Increased copper deposition in the placenta and fetal liver may adversely impact the fetus.
Maternal Adverse Reactions
Trientine may chelate non-copper cations (e.g., iron, calcium). Maintain appropriate levels during pregnancy [see Dosage and Administration (2.2), Warnings and Precautions (5.2, 5.3), and Drug Interactions (7.1)].
Data
Animal Data
In embryo-fetal development studies in rats, trientine was administered orally at doses up to approximately 835 mg/kg/day during organogenesis. An increased incidence of resorptions was observed at a dose lower than the maximum recommended dose (1,500 mg/day trientine free base equivalent), based on body surface area. An increased incidence of fetal abnormalities, including hemorrhage and edema, was observed at 2.7 times the maximum recommended dose, based on body surface area. When pregnant rats were treated with trientine and supplemented with copper (concentration of 50 mcg/g in the diet), fetal abnormalities were markedly reduced, indicating that the mechanism of embryo-fetal harm in rats with no copper supplementation was based on copper depletion (primary pharmacology of trientine).
In embryo-fetal development studies in mice, trientine dihydrochloride was administered orally at doses up to approximately 2,000 mg/kg/day during organogenesis. A dose-dependent increase in the incidence of abnormal fetuses occurred at all doses. Fetal abnormalities included hemorrhages and delayed ossification which were observed starting at 500 mg/kg/day, microcephaly and hydrocephaly starting at 1,000 mg/kg/day, and exencephaly at 2,000 mg/kg/day (the doses were 1.1, 2.1, and 4.3 times the maximum recommended dose, respectively, based on body surface area). The percentage of dams with total resorption was increased at 2,000 mg/kg/day (5.3 times the maximum recommended dose based on body surface area). The mechanism of embryo-fetal harm (e.g. copper depletion) was not determined in this study.
8.2 Lactation
Risk Summary
The available published data are inconsistent regarding the detection of trientine in breastmilk. Available published literature have not reported drug-associated adverse effects in infants exposed to trientine through breastmilk. There are no data on the effects of trientine on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CUVRIOR and any potential adverse effects on the breastfed infant from CUVRIOR or from the underlying condition.
8.4 Pediatric Use
The safety and effectiveness of CUVRIOR in pediatric patients have not been established.
8.5 Geriatric Use
In Trial 1, of the total number of CUVRIOR-treated patients, 1 (4%) was 65 years of age and older [see Clinical Studies (14)]. Clinical studies with trientine did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger adult patients. Other reported clinical experience with trientine is insufficient to determine differences in responses between geriatric and younger adult patients. In general, dose selection should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
10. Overdosage
Occasional cases of trientine overdose have been reported. A large overdose of 60 g of trientine hydrochloride (equivalent to 80 g CUVRIOR) resulted in nausea, vomiting, dizziness, mild acute kidney injury, mild hypophosphatemia, low serum zinc, and low serum copper. The patient recovered following intravenous hydration and supportive measures.
There is no antidote for trientine acute overdose.
Chronic use of trientine hydrochloride at dosages above the maximum recommended dosage has resulted in sideroblastic anemia.
11. Cuvrior Description
CUVRIOR contains trientine tetrahydrochloride which is a salt of trientine, a copper chelator. The structural formula of trientine tetrahydrochloride is:
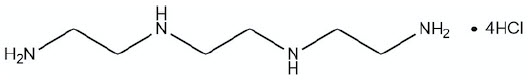
| Molecular Formula | C6H22Cl4N4 |
| Molecular Weight | 292.08 g mol-1 |
CUVRIOR (trientine tetrahydrochloride) tablets are for oral administration and contain 300 mg of trientine tetrahydrochloride (equivalent to 150 mg trientine). Tablets include the following inactive ingredients: colloidal silicon dioxide, glyceryl dibehenate, and mannitol. The film coating comprises ferric oxide yellow, glyceryl monocaprylocaprate (Type I), polyvinyl alcohol, purified talc, sodium lauryl sulfate, and titanium dioxide.
12. Cuvrior - Clinical Pharmacology
12.1 Mechanism of Action
Trientine, a copper chelator, eliminates absorbed copper from the body by forming a stable complex that is then eliminated through urinary excretion. Trientine also chelates copper in the intestinal tract, reducing copper absorption.
12.2 Pharmacodynamics
In Trial 1, serum non-ceruloplasmin copper (NCC) and 24-hour urinary copper excretion (UCE) were measured in adult patients with Wilson's disease who were de-coppered and tolerating penicillamine and who had switched to CUVRIOR. In patients who had switched to CUVRIOR, mean UCE decreased over time (see Table 4). On the other hand, the mean serum NCC in patients who had switched to CUVRIOR was comparable to the mean serum NCC in patients who remained on penicillamine [see Clinical Studies (14)].
| < 900 mg N=3 | 900 mg to 1,800 mg N=9 | 1,800 mg to 3,600 mg*
N=14 |
||||
|---|---|---|---|---|---|---|
| Visit | Serum NCC (mcg/L) | 24-hour UCE (mcg/24h) | Serum NCC (mcg/L) | 24-hour UCE (mcg/24h) | Serum NCC (mcg/L) | 24-hour UCE (mcg/24h) |
| NCC = non-ceruloplasmin copper (measured by an assay not commercially available); UCE = urinary copper excretion. Standard deviations are shown in parentheses. | ||||||
| Week 12† | 93.1 (78.2) | 438.0 (219.0) | 70.8 (20.3)‡ | 455.3 (283.4) | 62 .0 (25.2) | 612.8 (320.1)§ |
| Week 24 | 59.3 (32.5)¶ | 312.5 (174.7)¶ | 63.9 (19.6)‡ | 223.8 (134.3)‡ | 58.0 (15.6)# | 340.9 (160.9)# |
| Week 36 | 100.3 (81.8) | 425.6 (324.7) | 49.1 (12.3) | 267.8 (258.2) | 53.9 (9.6) | 274.0 (144.8) |
12.3 Pharmacokinetics
Absorption
The pharmacokinetic profile has been evaluated in healthy subjects following single oral administration of up to 1,500 mg CUVRIOR under fasting conditions.
The median time to the peak concentration (Tmax) of trientine ranged from 1.25 to 2 hours. Mean (± standard deviation) maximum plasma concentrations (Cmax) after 900 mg and 1,500 mg CUVRIOR were 2030±981, and 3430±1480 ng/mL, respectively. The systemic exposure (AUC) of trientine increased in a dose proportional manner over the range of 900 mg to 1,500 mg (mean AUCinf of 9750±4910 and 17200±9470 ng∙h/mL, respectively).
After administration of 900 mg (3 tablets) of CUVRIOR, the mean AUC was 11% lower compared to 750 mg (3 capsules) of trientine hydrochloride. After administration of 1,500 mg (5 tablets) of CUVRIOR, the mean AUC was comparable to 1,250 mg (5 capsules) of trientine hydrochloride [see Dosage and Administration (2.3)].
Elimination
The mean terminal half-life (t1/2) of trientine ranged from 13.8 to 16.5 hours.
Metabolism
Trientine is acetylated into two major active metabolites, N(1)-acetyltriethylenetetramine (MAT) and N(1),N(10)-diacetyltriethylenetetramine (DAT).
After the administration of a 900 mg dose of CUVRIOR, mean Cmax of MAT and DAT respectively were 1450±471 and 500±467 ng/mL and mean AUCinf was 15400±4200 and 5970±3390 ng∙h/mL. Mean T1/2 was 17.1±9.22 and 14.7±8.39 hours for MAT and DAT, respectively.
After the administration of a 1,500 mg dose of CUVRIOR, mean Cmax of MAT and DAT respectively were 2050±596 and 683±632 ng/mL and mean AUCinf was 22000±5770 and 7690±3970 ng∙h/mL. Mean T1/2 was 19.3±12.3 and 11.2±3.30 hours for MAT and DAT, respectively.
Specific Populations
Male and Female Patients
Following a single oral dose of 900 mg CUVRIOR, the systemic exposures (Cmax and AUCinf) were similar between males and females, although males appeared to have higher systemic exposures of trientine (approximately 30% and 50% higher for Cmax and AUCinf, respectively) following a single oral dose of 1,500 mg of CUVRIOR. This difference is not considered clinically meaningful for CUVRIOR.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
Trientine was positive in the bacterial reverse mutation (Ames) assay, the in vitro unscheduled DNA synthesis assay in rat hepatocytes, and the sister-chromatid exchange assay in Chinese hamster ovary (CHO) cells. Trientine was negative in the HGPRT gene mutation assay in CHO cells and the in vivo mouse micronucleus assay.
13.2 Animal Toxicology and/or Pharmacology
Severe pulmonary toxicity occurred in a 26-week oral toxicity study of trientine dihydrochloride in rats. However, the relevance of this finding to safety risk in patients with Wilson's disease is unknown. Deaths related to pulmonary injury occurred in male rats at 175 and 600 mg/kg/day (1/12 and 3/20 males, respectively). Pulmonary lesions were observed in male rats at 50 mg/kg/day (lower than the maximum recommended dose [1,500 mg/day trientine free base equivalent] based on body surface area). In female rats, pulmonary lesions occurred at 175 mg/kg/day (lower than the maximum recommended dose based on body surface area). Pulmonary toxicity was dose-dependent in both sexes. The lesions included alveolar fibrosis, bronchiolar hyperplasia/ epithelialization of alveolar wall, focal chronic interstitial pneumonitis, bronchiolar epithelium hypertrophy, alveolar hemorrhage (males only), necrosis/regenerative hyperplasia of terminal bronchioles (males only), and acute interstitial pneumonitis (males only). Most of the pulmonary lesions remained after a 13-week recovery period, and were considered as irreversible (e.g., alveolar fibrosis, bronchiolar hyperplasia/epithelialization of alveolar wall, focal chronic interstitial pneumonitis). Pulmonary toxicity also occurred in a 13-week oral study of trientine dihydrochloride in mice. The effects included interstitial inflammation and alveolar histiocytic infiltration, observed at a dose approximately equal to the maximum recommended dose based on body surface area. The mechanism of pulmonary toxicity was not clearly established as due to copper depletion (primary pharmacology) or due to other drug activity.
Neurological and/or musculoskeletal clinical signs (abnormal "stiff-legged" gait, limited use of limb, underactivity, body tremors) occurred in a 26-week oral toxicity study of trientine dihydrochloride in dogs at 5.5 times the estimated human exposure at the maximum recommended dose (based on AUC). The mechanism for these clinical signs was not clearly established as due to copper depletion (primary pharmacology) or due to other drug activity. Because of the severity of the clinical signs at exposures above 5.5 times the maximum exposure in humans, some dogs were sacrificed for humane reasons after 9 weeks of treatment, and treatment for the remaining animals in the same dose group was stopped after 10 weeks. Neurological and/or musculoskeletal clinical signs were generally not observed at exposures that were 3.4 times the human exposure at the maximum recommended dose.
14. Clinical Studies
The effectiveness of CUVRIOR for the treatment of adult patients with stable Wilson's disease who are decoppered and tolerant to penicillamine was demonstrated in a phase 3 trial (Trial 1). In addition, the safety and effectiveness of CUVRIOR in Wilson's Disease is further supported by studies of another trientine product in patients intolerant to penicillamine.
Trial 1 was a randomized, active-controlled, multi-center, non-inferiority study (NCT03539952) conducted in 53 adult patients with Wilson's disease. The objective was to compare treatment with CUVRIOR to treatment with penicillamine. All patients had been receiving penicillamine for at least 1 year prior to study entry, were adequately controlled and tolerating penicillamine, and had a serum NCC level between ≥ 25 and ≤ 150 mcg/L.
At the start of the study, patients entered a 12-week baseline period and continued to receive their established total daily dosage of penicillamine for 12 weeks. At Week 12, patients were randomized to either remain on penicillamine (N=27) or to switch to CUVRIOR (N=26) for the 24-week post-randomization period (i.e., Week 12 through Week 36). For patients switching to CUVRIOR, where possible, the initial total daily dosage was determined as the trientine base in mg that was the same as the patient's total daily dosage in mg of penicillamine. Where a direct mg to mg conversion was not possible, the total daily dosage of CUVRIOR was rounded to the nearest 150 mg of trientine base (300 mg trientine tetrahydrochloride salt) to the penicillamine total daily dosage. The dosage was permitted to be adjusted depending on clinical response. The mean CUVRIOR total daily dosage was 1,800 mg. Upon switching from penicillamine to CUVRIOR, 3 patients switched to a CUVRIOR total daily dosage < 900 mg, 9 patients to a total daily dosage between 900 mg and 1,800 mg, and 14 patients to a total daily dosage of 1,800 mg or greater. Three out of 26 patients increased and one patient reduced the total daily dosage across the 24-week post-randomization period.
The results are presented in Table 5. The primary efficacy endpoint was the mean serum non-ceruloplasmin copper (NCC) level at 24 weeks post-randomization (Week 36). At Week 12 (prior to initiation of randomized treatment), the mean (95% CI) NCC levels in the penicillamine and CUVRIOR arms were 77 mcg/L (66; 88) and 66 mcg/L (55; 76), respectively. The mean NCC level at Week 36 as measured using an assay not commercially available was similar in patients receiving CUVRIOR and in patients receiving penicillamine. However, the mean 24-hour urinary copper excretion (UCE) at Week 36 was lower in patients receiving CUVRIOR as compared to patients receiving penicillamine. A decrease in UCE has been observed when switching patients from penicillamine products to trientine products. All patients in both treatment arms were considered clinically stable as determined by an adjudication committee at Week 36.
| Parameter | Penicillamine Arm (N=27) | CUVRIOR Arm (N=26) | Difference† |
|---|---|---|---|
| CI = confidence interval; NCC = non-ceruloplasmin copper; UCE = urinary copper excretion | |||
| Serum NCC (mcg/L) | |||
| Mean‡ (95% CI) | 46 (35; 58) | 56 (44; 67) | -9 (-24; 6) |
| 24-hour UCE (mcg/24h) | |||
| Mean‡ (95% CI) | 511 (415; 607) | 274 (183; 366) | 236 (111; 361) |
16. How is Cuvrior supplied
CUVRIOR tablets, 300 mg of trientine tetrahydrochloride, are oblong, yellow coated, functionally scored, and imprinted with OL75 on each side. Each large carton (NDC 81802-001-72) contains nine child-resistant small cartons (NDC 81802-001-08), each containing a blister pack of 8 tablets (a total of 72 tablets in the large carton). The fewest number of tablets that can be dispensed is 8 tablets in a small carton.
Do not remove tablets from the blister pack until the time of dosing.
17. Patient Counseling Information
Administration Instructions
Advise patients or their caretaker(s) to [see Dosage and Administration (2.2)]:
- Take CUVRIOR on an empty stomach (at least 1 hour before meals or 2 hours after meals and at least 1 hour apart from other food or milk).
- Avoid concomitant use of mineral supplements (e.g. iron, zinc, calcium, magnesium). If concomitant use of mineral supplements is unavoidable [see Drug Interactions (7.1)]:
- Advise patients taking an iron supplement to take CUVRIOR at least 2 hours before or 2 hours after taking an iron supplement.
- Advise patients taking other mineral supplements to take CUVRIOR at least 1 hour before or 2 hours after taking other mineral supplements.
- Take CUVRIOR at least 1 hour apart from any other oral drug.
- Remove CUVRIOR tablets from the blister pack only at the time of dosing.
- Swallow tablets of CUVRIOR without crushing, chewing, or dissolving tablets. Advise patients that if they have difficulty swallowing the tablet whole, the scored tablet can be divided into two equal halves. Advise patients not to store the tablet for future use after the blister has been opened.
Laboratory Monitoring
Advise patients that copper level monitoring is required periodically during CUVRIOR therapy [see Dosage and Administration (2.4) and Warnings and Precautions (5.1, 5.2)].
Potential for Worsening of Clinical Symptoms at Initiation of Therapy
Advise patients or their caretaker(s) to notify their healthcare provider if they experience worsening of clinical symptoms, including neurological deterioration [see Warnings and Precautions (5.1)].
Copper or Iron Deficiency
Advise patients or their caretaker(s) to notify their healthcare provider if they experience any signs or symptoms of copper or iron deficiency [see Warnings and Precautions (5.2, 5.3)].
Hypersensitivity Reactions
Advise patients or their caretaker(s) to notify their healthcare provider if they develop a rash or other hypersensitivity reaction while taking CUVRIOR [see Warnings and Precautions (5.4)].

| CUVRIOR
trientine tetrahydrochloride tablet, film coated |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Orphalan SA (263303972) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Delpharm Evreux | 573370603 | MANUFACTURE(81802-001) , PACK(81802-001) , ANALYSIS(81802-001) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Delpharm Lille | 283582547 | MANUFACTURE(81802-001) , PACK(81802-001) , ANALYSIS(81802-001) | |
More about Cuvrior (trientine)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Imprints, shape & color data
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: chelating agents
- Breastfeeding
- En español