Aduhelm: Package Insert / Prescribing Info
Package insert / product label
Generic name: aducanumab
Dosage form: injection, solution
Drug class: Miscellaneous central nervous system agents
Medically reviewed by Drugs.com. Last updated on Sep 10, 2023.
The Aduhelm brand name has been discontinued in the U.S. If generic versions of this product have been approved by the FDA, there may be generic equivalents available.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
ADUHELM® (aducanumab-avwa) injection, for intravenous use
Initial U.S. Approval: 2021
WARNING: AMYLOID RELATED IMAGING ABNORMALITIES
See full prescribing information for complete boxed warning.
Monoclonal antibodies directed against aggregated forms of beta amyloid, including ADUHELM, can cause amyloid related imaging abnormalities (ARIA), characterized as ARIA with edema (ARIA-E) and ARIA with hemosiderin deposition (ARIA-H). ARIA is usually asymptomatic, although rarely serious and life-threatening events can occur. Serious intracerebral hemorrhage greater than 1 cm have occurred in patients treated with this class of medications. (5.1, 6.1)
ApoE ε4 Homozygotes
Patients treated with this class of medications, including ADUHELM, who are ApoE ε4 homozygotes have a higher incidence of ARIA, including symptomatic and serious ARIA, compared to heterozygotes and noncarriers. Testing for ApoE ε4 status should be performed prior to initiation of treatment to inform the risk of developing ARIA. Prior to testing, prescribers should discuss with patients the risk of ARIA across genotypes and the implications of genetic testing results. (5.1)
Consider the benefit of ADUHELM for the treatment of Alzheimer's disease and potential risk of serious adverse events associated with ARIA when deciding to initiate treatment with ADUHELM. (5.1, 14)
Recent Major Changes
Indications and Usage for Aduhelm
ADUHELM is an amyloid beta-directed antibody indicated for the treatment of Alzheimer's disease. Treatment with ADUHELM should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. This indication is approved under accelerated approval based on reduction in amyloid beta plaques observed in patients treated with ADUHELM. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trial(s). (1)
Aduhelm Dosage and Administration
- Confirm the presence of amyloid beta pathology prior to initiating treatment. (2.1)
- Titration is required for treatment initiation. (2.2 )
- The recommended maintenance dosage is 10 mg/kg administered as an intravenous infusion over approximately one hour every four weeks. (2.2 )
- Obtain a recent (within one year) brain MRI prior to initiating treatment. (2.3, 5.1)
- Obtain MRIs prior to the 5th, 7th, 9th, and 12th infusions; if radiographically observed ARIA occurs, treatment recommendations are based on type, severity, and presence of symptoms. (2.3, 5.1)
- Dilution in 100 mL of 0.9% Sodium Chloride Injection, USP, is required prior to administration. (2.4)
- Administer as an intravenous infusion over approximately one hour via a 0.2 or 0.22 micron in-line filter. (2.5)
Dosage Forms and Strengths
Contraindications
None. (4)
Warnings and Precautions
- Amyloid Related Imaging Abnormalities (ARIA): Enhanced clinical vigilance for ARIA is recommended during the first 8 doses of treatment with ADUHELM, particularly during titration. Risk of ARIA, including symptomatic ARIA, was increased in apolipoprotein E ε4 homozygotes compared to heterozygotes and noncarriers. If a patient experiences symptoms which could be suggestive of ARIA, clinical evaluation should be performed, including MRI testing if indicated. (2.3, 5.1)
- Hypersensitivity Reactions: Angioedema and urticaria have occurred. If a hypersensitivity reaction occurs, promptly discontinue the infusion of ADUHELM and initiate appropriate therapy. (5.2)
Adverse Reactions/Side Effects
Most common adverse reactions (at least 10% and higher incidence compared to placebo): ARIA-Edema, headache, ARIA-H microhemorrhage, ARIA-H superficial siderosis, and fall. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Biogen at 1-833-425-9360 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 8/2023
Full Prescribing Information
WARNING: AMYLOID RELATED IMAGING ABNORMALITIES
Monoclonal antibodies directed against aggregated forms of beta amyloid, including ADUHELM, can cause amyloid related imaging abnormalities (ARIA), characterized as ARIA with edema (ARIA-E) and ARIA with hemosiderin deposition (ARIA-H). Incidence and timing of ARIA vary among treatments. ARIA usually occurs early in treatment and is usually asymptomatic, although serious and life-threatening events rarely can occur. Serious intracerebral hemorrhages, some of which have been fatal, have been observed in patients treated with this class of medications [see Warnings and Precautions (5.1), Adverse Reactions (6.1)].
ApoE ε4 Homozygotes
Patients who are apolipoprotein E ε4 (ApoE ε4) homozygotes (approximately 15% of Alzheimer's disease patients) treated with this class of medications, including ADUHELM, have a higher incidence of ARIA, including symptomatic, serious, and severe radiographic ARIA, compared to heterozygotes and noncarriers. Testing for ApoE ε4 status should be performed prior to initiation of treatment to inform the risk of developing ARIA. Prior to testing, prescribers should discuss with patients the risk of ARIA across genotypes and the implications of genetic testing results. Prescribers should inform patients that if genotype testing is not performed they can still be treated with ADUHELM; however, it cannot be determined if they are ApoE ε4 homozygotes and at higher risk for ARIA [see Warnings and Precautions (5.1)].
Consider the benefit of ADUHELM for the treatment of Alzheimer's disease and potential risk of serious adverse events associated with ARIA when deciding to initiate treatment with ADUHELM [see Warnings and Precautions (5.1) and Clinical Studies (14)].
1. Indications and Usage for Aduhelm
ADUHELM is indicated for the treatment of Alzheimer's disease. Treatment with ADUHELM should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. This indication is approved under accelerated approval based on reduction in amyloid beta plaques observed in patients treated with ADUHELM [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trial(s).
2. Aduhelm Dosage and Administration
2.1 Patient Selection
Confirm the presence of amyloid beta pathology prior to initiating treatment [see Clinical Pharmacology (12.1)].
2.2 Dosing Instructions
After an initial titration, the recommended dosage of ADUHELM is 10 mg/kg (see Table 1). ADUHELM must be diluted and is administered as an intravenous infusion over approximately one hour every four weeks.
| Intravenous Infusion
(every 4 weeks) | ADUHELM Dosage (administered over approximately one hour) |
| Infusion 1 and 2 | 1 mg/kg |
| Infusion 3 and 4 | 3 mg/kg |
| Infusion 5 and 6 | 6 mg/kg |
| Infusion 7 and beyond | 10 mg/kg |
If an infusion is missed, resume administration at the same dose as soon as possible and at least 21 days apart.
2.3 Monitoring and Dosing Interruptions for Amyloid Related Imaging Abnormalities
ADUHELM can cause amyloid related imaging abnormalities -edema (ARIA-E) and hemosiderin deposition (ARIA-H) [see Warnings and Precautions (5.1)].
Monitoring for ARIA
Obtain a recent brain magnetic resonance imaging (MRI) prior to initiating treatment with ADUHELM. Obtain MRIs prior to the 5th infusion (first dose of 6 mg/kg), 7th infusion (first dose of 10 mg/kg), 9th infusion (third dose of 10 mg/kg), and 12th infusion (sixth dose of 10 mg/kg). If a patient experiences symptoms suggestive of ARIA, clinical evaluation should be performed, including an MRI if indicated.
Recommendations for Dosing Interruptions in Patients with ARIA
If dosing is resumed following a temporary suspension, dosing may resume at that same dose and titration schedule prior to the dosing suspension. The benefits of reaching and maintaining the 10 mg/kg dosage should be considered when evaluating a potential dose suspension.
ARIA-E
The recommendations for dosing interruptions for patients with ARIA-E are provided in Table 2.
|
1 Mild: discomfort noticed, but no disruption of normal daily activity. |
|||
|
2 Suspend until MRI demonstrates radiographic resolution and symptoms, if present, resolve; consider a follow-up MRI to assess for resolution 2 to 4 months after initial identification. Resumption of dosing should be guided by clinical judgment. |
|||
|
3 See table 4. |
|||
| Clinical Symptom Severity1 | ARIA-E Severity on MRI3 | ||
| Mild | Moderate | Severe | |
| Asymptomatic | May continue dosing at current dose and schedule | Suspend dosing2 | Suspend dosing2 |
| Mild
| May continue dosing based on clinical judgment | Suspend dosing2 | |
| Moderate or Severe
| Suspend dosing2 | ||
ARIA-H
The recommendations for dosing interruptions for patients with ARIA-H are provided in Table 3.
|
1 Suspend until MRI demonstrates radiographic stabilization and symptoms, if present, resolve; resumption of dosing should be guided by clinical judgment; consider a follow-up MRI to assess for stabilization 2 to 4 months after initial identification. |
|||
|
2 Suspend until MRI demonstrates radiographic stabilization and symptoms, if present, resolve; use clinical judgment in considering whether to continue treatment or permanently discontinue ADUHELM. |
|||
|
3 See table 4. |
|||
| Clinical Symptom Severity | ARIA-H Severity on MRI3 | ||
| Mild | Moderate | Severe | |
| Asymptomatic | May continue dosing at current dose and schedule | Suspend dosing1 | Suspend dosing2 |
| Symptomatic
| Suspend dosing1 | Suspend dosing1 | |
In patients who develop intracerebral hemorrhage greater than 1 cm in diameter during treatment with ADUHELM, suspend dosing until MRI demonstrates radiographic stabilization and symptoms, if present, resolve. In Studies 1 and 2, dosing was permanently discontinued in patients who developed intracerebral hemorrhage greater than 1 cm in diameter. Use clinical judgment in considering whether to continue treatment after radiographic stabilization and resolution of symptoms or permanently discontinue ADUHELM.
2.4 Dilution Instructions
- Prior to administration, ADUHELM must be diluted in 100 mL of 0.9% Sodium Chloride Injection, USP.
- Use aseptic technique when preparing the ADUHELM diluted solution for intravenous infusion.
- Calculate the dose, total volume of ADUHELM solution required, and the number of vials needed based on the patient's actual body weight. Each vial contains an ADUHELM concentration of 100 mg per mL.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Check that the ADUHELM solution is clear to opalescent and colorless to yellow solution. Do not use if opaque particles, discoloration, or other foreign particles are present.
- Remove the flip-off cap from the vial. Insert the syringe needle into the vial through the center of the rubber stopper.
- Withdraw the required volume of ADUHELM from the vial(s) and add to an infusion bag of 100 mL of 0.9% Sodium Chloride Injection, USP. Do not use other intravenous diluents to prepare the ADUHELM diluted solution.
- Each vial is for single-dose only. Discard any unused portion.
- Gently invert the infusion bag containing the ADUHELM diluted solution to mix completely. Do not shake.
- After dilution, immediate use is recommended. If not administered immediately, store the diluted solution of ADUHELM in 0.9% Sodium Chloride Injection, USP refrigerated at 2°C to 8°C (36°F to 46°F) for up to 3 days, or at room temperature up to 30°C (86°F) for up to 12 hours. Do not freeze.
2.5 Administration Instructions
- Visually inspect the ADUHELM diluted solution for particles or discoloration prior to administration. Do not use if it is discolored, or opaque or foreign particles are seen.
- Prior to infusion, allow the ADUHELM diluted solution to warm to room temperature.
- Infuse ADUHELM diluted solution intravenously over approximately one hour through an intravenous line containing a sterile, low-protein binding, 0.2 or 0.22 micron in-line filter.
- Promptly discontinue the infusion upon the first observation of any signs or symptoms consistent with a hypersensitivity-type reaction [see Warnings and Precautions (5.2)].
3. Dosage Forms and Strengths
ADUHELM is a clear to opalescent and colorless to yellow solution, available as:
- Injection: 170 mg/1.7 mL (100 mg/mL) in a single-dose vial
- Injection: 300 mg/3 mL (100 mg/mL) in a single-dose vial
5. Warnings and Precautions
5.1 Amyloid Related Imaging Abnormalities
Monoclonal antibodies directed against aggregated forms of beta amyloid, including ADUHELM, can cause amyloid related imaging abnormalities (ARIA), characterized as ARIA with edema (ARIA-E), which can be observed on MRI as brain edema or sulcal effusions, and ARIA with hemosiderin deposition (ARIA-H), which includes microhemorrhage and superficial siderosis. ARIA can occur spontaneously in patients with Alzheimer's disease. ARIA-H associated with monoclonal antibodies directed against aggregated forms of beta amyloid generally occurs in association with an occurrence of ARIA-E. ARIA-H of any cause and ARIA-E can occur together.
ARIA usually occurs early in treatment and is usually asymptomatic, although serious and life-threatening events, including seizure and status epilepticus, rarely can occur. When present, reported symptoms associated with ARIA may include headache, confusion, visual changes, dizziness, nausea, and gait difficulty. Focal neurologic deficits may also occur. Symptoms associated with ARIA usually resolve over time. The risk of ARIA, including symptomatic and serious ARIA, is increased in apolipoprotein E ε4 (ApoE ε4) homozygotes. In addition to ARIA, intracerebral hemorrhages greater than 1 cm in diameter have occurred in patients treated with ADUHELM.
Consider the benefit of ADUHELM for the treatment of Alzheimer's disease and potential risk of serious adverse events associated with ARIA when deciding to initiate treatment with ADUHELM.
Incidence of ARIA
Symptomatic ARIA occurred in 10% (110/1105) of patients treated with ADUHELM in Studies 1 and 2. Serious symptoms associated with ARIA were reported in 0.3% of patients treated with ADUHELM. Clinical symptoms associated with ARIA resolved in 88% of patients during the period of observation. Overall, recurrent episodes of ARIA-E were less frequently symptomatic (12%) compared with initial episodes of ARIA-E (25%).
Including asymptomatic radiographic events, ARIA was observed in 41% (454/1105) of patients treated with ADUHELM 10mg/kg compared to 10% (111/1087) of patients on placebo in Studies 1 and 2.
ARIA-E was observed in 35% (387/1105) of patients treated with ADUHELM 10 mg/kg compared with 3% (29/1087) of patients on placebo. ARIA-H was observed in 28% (312/1105) of patients treated with ADUHELM compared to 9% (94/1087) of patients on placebo. There was no increase in isolated ARIA-H (i.e., ARIA-H in patients who did not also experience ARIA-E) for ADUHELM compared to placebo.
The overall incidence of seizure, independent of ARIA, was 0.5% in the 10 mg/kg ADUHELM group and 0.8% in the placebo group in Studies 1 and 2. In patients with ARIA in the 10 mg/kg ADUHELM group, the incidence of seizure was 0.7%. Status epilepticus was reported in the placebo-controlled and long-term extension studies in patients treated with ADUHELM.
ApoE ε4 Carrier Status and Risk of ARIA
Approximately 15% of Alzheimer's disease patients are ApoE ε4 homozygotes. In Studies 1 and 2, among patients with a known ApoE ε4 genotype, 17% (182/1103) of patients in the ADUHELM group were ApoE ε4 homozygotes, 51% (564/1103) were heterozygotes, and 32% (357/1103) were noncarriers. The incidence of symptomatic ARIA was higher in ApoE ε4 homozygotes (16%) than in heterozygotes (11%) and noncarriers (5%) among patients treated with ADUHELM. However, the incidence of serious adverse reactions with ARIA-E, including risk of death, persistent or significant disability or incapacity, hospitalization, or other medically important event that may require intervention to prevent serious outcomes, was similar for ApoE ε4 carriers and noncarriers (2% in homozygotes, 1% in heterozygotes, 2% in noncarriers). The recommendations on management of ARIA do not differ between ApoE ε4 carriers and noncarriers [see Dosage and Administration (2.3)]. Testing for ApoE ε4 status should be performed prior to initiation of treatment to inform the risk of developing ARIA. Prior to testing, prescribers should discuss with patients the risk of ARIA across genotypes and the implications of genetic testing results. Prescribers should inform patients that if genotype testing is not performed they can still be treated with ADUHELM; however, it cannot be determined if they are ApoE ε4 homozygotes and at a higher risk for ARIA. An FDA-authorized test for detection of ApoE ε4 alleles to identify patients at risk of ARIA if treated with ADUHELM is not currently available. Currently available tests used to identify ApoE ε4 alleles may vary in accurary and design.
Radiographic Findings
The radiographic severity of ARIA associated with ADUHELM was classified by the criteria shown in Table 4.
| ARIA Type | Radiographic Severity | ||
| Mild | Moderate | Severe | |
| ARIA-E | FLAIR hyperintensity confined to sulcus and or cortex/subcortical white matter in one location < 5 cm | FLAIR hyperintensity 5 to 10 cm, or more than 1 site of involvement, each measuring < 10 cm | FLAIR hyperintensity measuring > 10 cm, often with significant subcortical white matter and/or sulcal involvement. One or more separate sites of involvement may be noted. |
| ARIA-H microhemorrhage | ≤ 4 new incident microhemorrhages | 5 to 9 new incident microhemorrhages | 10 or more new incident microhemorrhages |
| ARIA-H superficial siderosis | 1 focal area of superficial siderosis | 2 focal areas of superficial siderosis | > 2 focal areas of superficial siderosis |
The majority of ARIA-E radiographic events occurred early in treatment (within the first 8 doses), although ARIA can occur at any time and patients can have more than 1 episode. The maximum radiographic severity of ARIA-E in patients treated with ADUHELM was mild in 10% (115/1105) of patients, moderate in 20% (223/1105) of patients, and severe in 4% (49/1105) of patients. Resolution on MRI occurred in 68% of ARIA-E patients by 12 weeks, 91% by 20 weeks, and 98% overall after detection. The maximum radiographic severity of ARIA-H microhemorrhage in patients treated with ADUHELM was mild in 14% (154/1105) of patients, moderate in 3% (29/1105) of patients, and severe in 3% (29/1105) patients. The maximum radiographic severity of ARIA-H superficial siderosis in patients treated with ADUHELM was mild in 7% (79/1105) of patients, moderate in 4% (47/1105) of patients, and severe in 3% (36/1105) of patients. Among patients treated with ADUHELM, the incidence of severe radiographic ARIA-E was highest in ApoE ε4 homozygotes 11% (20/182), compared to heterozygotes 4% (21/564) or noncarriers 2% (8/357). Among patients treated with ADUHELM, the incidence of severe radiographic ARIA-H (microhemorrhage or superficial siderosis) was highest in ApoE ε4 homozygotes 20% (36/182), compared to heterozygotes 4% (21/564) or noncarriers 2% (6/357).
Intracerebral Hemorrhage
Intracerebral hemorrhage greater than 1 cm in diameter was reported in 0.5% (6/1105) of patients in Studies 1 and 2 after treatment with ADUHELM compared to 0.4% (4/1087) of patients on placebo. Fatal events of intracerebral hemorrhage in patients taking ADUHELM have been observed.
Concomitant Antithrombotic Medication
Patients were excluded from enrollment in Studies 1 and 2 for use of antiplatelet or anticoagulant medications other than 325 mg or less daily of aspirin. Although patients were allowed to receive aspirin in daily doses of 325 mg or less, some patients, because of intercurrent medical events that occurred after enrollment and required treatment, received aspirin in doses greater than 325 mg, other antiplatelet drugs, or anticoagulants during Studies 1 and 2. Patients who received ADUHELM and an antithrombotic medication (aspirin, other antiplatelets, or anticoagulants ) did not have an increased risk of ARIA-H or intracerebral hemorrhage compared to patients who received placebo and an antithrombotic medication. The majority of exposures to antithrombotic medications were to aspirin; few patients were exposed to other antiplatelet drugs or anticoagulants, limiting any meaningful conclusions about the risk of ARIA or intracerebral hemorrhage in patients taking other antiplatelet drugs or anticoagulants.
Because intracerebral hemorrhages greater than 1 cm in diameter have been observed in patients taking ADUHELM, additional caution should be exercised when considering the administration of anticoagulants or a thrombolytic agent (e.g., tissue plasminogen activator) to a patient already being treated with ADUHELM.
Other Risk Factors for Intracerebral Hemorrhage
Patients were excluded from enrollment in Studies 1 and 2 for findings on neuroimaging that indicated an increased risk for intracerebral hemorrhage. These included findings suggestive of cerebral amyloid angiopathy (prior intracerebral hemorrhage greater than 1 cm in diameter, more than 4 microhemorrhages, superficial siderosis, and history of diffuse white matter disease). Vasogenic edema could also be suggestive of cerebral amyloid angiopathy. These and other lesions (aneurysm, vascular malformation) could potentially increase the risk of intracerebral hemorrhage.
The presence of an ApoE ε4 allele is also associated with cerebral amyloid angiopathy which has an increased risk for intracerebral hemorrhage.
Caution should be exercised when considering the use of ADUHELM in patients with factors that indicate an increased risk for intracerebral hemorrhage and in particular for patients who need to be on anticoagulant therapy.
Monitoring and Dose Management Guidelines
Recommendations for dosing in patients with ARIA-E depend on clinical symptoms and radiographic severity [see Dosage and Administration (2.3)]. Recommendations for dosing in patients with ARIA-H depend on the type of ARIA-H and radiographic severity [see Dosage and Administration (2.3)]. Use clinical judgment in considering whether to continue dosing in patients with recurrent ARIA-E.
Baseline brain MRI and periodic monitoring with MRI are recommended [see Dosage and Administration (2.3)]. Enhanced clinical vigilance for ARIA is recommended during the first 8 doses of treatment with ADUHELM, particularly during titration. If a patient experiences symptoms suggestive of ARIA, clinical evaluation should be performed, including MRI if indicated. If ARIA is observed on MRI, careful clinical evaluation should be performed prior to continuing treatment.
There is limited experience in patients who continued dosing through symptomatic ARIA-E or through asymptomatic moderate or severe ARIA-E. There are limited data in dosing patients who experienced recurrent ARIA-E.
The Alzheimer's Network for Treatment and Diagnostics (ALZ-NET) is a voluntary provider-enrolled patient registry that collects information on treatments for Alzheimer's disease, including ADUHELM. Providers may obtain information about the registry at www.alz-net.org or contact alz-net@acr.org.
5.2 Hypersensitivity Reactions
Angioedema and urticaria were reported in one patient in the placebo-controlled period of Studies 1 and 2, and occurred during the ADUHELM infusion. Promptly discontinue the infusion upon the first observation of any signs or symptoms consistent with a hypersensitivity reaction, and initiate appropriate therapy.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Amyloid Related Imaging Abnormalities [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of ADUHELM has been evaluated in 3,078 patients who received at least one dose of ADUHELM. In two placebo-controlled studies (Studies 1 and 2) in patients with Alzheimer's disease, a total of 1105 patients received ADUHELM 10 mg/kg [see Clinical Studies (14)]. Of these 1105 patients, approximately 52% were female, 76% were White, 10% were Asian, and 3% were of Hispanic or Latino ethnicity. The mean age at study entry was 70 years (range from 50 to 85).
In the combined placebo-controlled and long-term extension periods of Studies 1 and 2, 834 patients received at least one dose of ADUHELM 10 mg/kg once monthly for at least 6 months, 551 patients for at least 12 months, and 309 patients for at least 18 months. In the combined placebo-controlled and long-term extension periods, 5% (66 out of 1386) of patients in the 10 mg/kg dose group withdrew from the study because of an adverse reaction. The most common adverse reaction resulting in study withdrawal in the combined placebo-controlled and long-term extension periods was ARIA-H superficial siderosis. Table 5 shows adverse reactions that were reported in at least 2% of patients treated with ADUHELM and at least 2% more frequently than in patients on placebo.
| Adverse Reaction | ADUHELM
10 mg/kg N=1105 % | Placebo
N=1087 % |
|---|---|---|
|
aHeadache includes the adverse reaction related terms headache, head discomfort, migraine, migraine with aura, and occipital neuralgia. |
||
|
bDiarrhea includes the adverse reaction related terms diarrhea and infectious diarrhea. |
||
|
cConfusion/Delirium/Altered Mental Status/Disorientation includes the adverse reaction related terms confusional state, delirium, altered state of consciousness, disorientation, depressed level of consciousness, disturbance in attention, mental impairment, mental status changes, postoperative confusion, and somnolence. |
||
| ARIA-E | 35 | 3 |
| Headachea | 21 | 16 |
| ARIA-H microhemorrhage | 19 | 7 |
| ARIA-H superficial siderosis | 15 | 2 |
| Fall | 15 | 12 |
| Diarrheab | 9 | 7 |
| Confusion/Delirium/Altered Mental Status/Disorientationc | 8 | 4 |
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate data on ADUHELM use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Intravenous administration of aducanumab-avwa (0, 100, 300, or 1000 mg/kg/week) to female rats through organogenesis had no adverse effect on embryofetal development.
Intravenous administration of aducanumab-avwa (0, 100, 300, or 1000 mg/kg/week) to female rats throughout pregnancy and lactation had no adverse effects on pre- or postnatal development.
The relevance of these data to humans is limited because aggregated amyloid beta, the pharmacological target of aducanumab-avwa, is not present in rat.
8.2 Lactation
Risk Summary
There are no data on the presence of aducanumab-avwa in human milk, the effects on the breastfed infant, or the effects of the drug on milk production. Published data from other monoclonal antibodies generally indicate low passage of monoclonal antibodies into human milk and limited systemic exposure in the breastfed infant. The effects of this limited exposure are unknown. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ADUHELM and any potential adverse effects on the breastfed infant from ADUHELM or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of ADUHELM in pediatric patients have not been established.
8.5 Geriatric Use
In Studies 1 and 2, the age of patients ranged from 50 to 85 years, with a mean age of 70 years; 79% were 65 and older, and 32% were 75 and older. There were no notable differences in the incidence of adverse reactions between these age groups, and no additional safety concerns in patients 65 years of age and older compared to younger patients.
11. Aduhelm Description
Aducanumab-avwa is a recombinant human immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble and insoluble forms of amyloid beta, and is expressed in a Chinese hamster ovary cell line. Aducanumab-avwa has an approximate molecular weight of 146 kDa.
ADUHELM (aducanumab-avwa) injection is a preservative-free, sterile, clear to opalescent, and colorless to yellow solution for intravenous infusion after dilution supplied in single-dose vials available in concentrations of 170 mg/1.7 mL (100 mg/mL) or 300 mg/3 mL (100 mg/mL) of ADUHELM.
Each mL of solution contains 100 mg of aducanumab-avwa and L-arginine hydrochloride (31.60 mg), L-histidine (0.60 mg), L-histidine hydrochloride monohydrate (3.39 mg), L-methionine (1.49 mg), polysorbate 80 (0.50 mg), and Water for Injection at an approximate pH of 5.5.
12. Aduhelm - Clinical Pharmacology
12.1 Mechanism of Action
Aducanumab-avwa is a human, immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble and insoluble forms of amyloid beta. The accumulation of amyloid beta plaques in the brain is a defining pathophysiological feature of Alzheimer's disease. ADUHELM reduces amyloid beta plaques, as evaluated in Studies 1, 2, and 3 [see Clinical Studies (14)].
12.2 Pharmacodynamics
Effect of ADUHELM on Amyloid Beta Pathology
ADUHELM reduced amyloid beta plaque in a dose- and time-dependent manner in Study 1, Study 2, and Study 3, compared with placebo [see Dosage and Administration (2.2) and Clinical Studies (14)].
The effect of ADUHELM on amyloid beta plaque levels in the brain was evaluated using PET imaging (18F-florbetapir tracer). The PET signal was quantified using the Standard Uptake Value Ratio (SUVR) method to estimate brain levels of amyloid beta plaque in composites of brain areas expected to be widely affected by Alzheimer's disease pathology (frontal, parietal, lateral temporal, sensorimotor, and anterior and posterior cingulate cortices), compared to a brain region expected to be spared of such pathology (cerebellum). The SUVR was also expressed on the Centiloid scale.
In substudies of Study 1 and Study 2, ADUHELM reduced amyloid beta plaque levels in the brain, producing reductions at both ADUHELM low dose and high dose levels and at both Weeks 26 and 78 (p < 0.0001), compared to placebo. The magnitude of reduction was time- and dose-dependent. In the long-term extension of Study 1 and Study 2, a continued decrease in brain amyloid beta plaque levels was observed at Week 132 in patients initially randomized to ADUHELM.
In Study 3, ADUHELM reduced amyloid beta plaque levels in the brain, producing statistically significant dose- and time-dependent reductions compared to placebo in the 3 mg/kg, 6 mg/kg, and 10 mg/kg ADUHELM treatment groups at Week 26, and in all ADUHELM treatment groups at Week 54. Among those dosed with ADUHELM during the placebo-controlled period in Study 3, amyloid beta plaque levels in the brain continued to decline in a time- and dose-dependent manner in the long-term extension period through Week 222.
Effect of ADUHELM on Tau Pathophysiology
ADUHELM reduced markers of tau pathophysiology (CSF p-Tau 181, plasma p-Tau 181, and Tau PET) and neurodegeneration (CSF t-Tau) in Study 1 and Study 2 [see Clinical Studies (14)].
ADUHELM reduced CSF levels of p-Tau 181 in substudies conducted in Study 1 and Study 2. The adjusted mean change from baseline in CSF p-Tau 181 levels relative to placebo was in favor of the ADUHELM low (p<0.01) and high (p<0.001) dose groups at Week 78 in Study 1. Results in Study 2 numerically favored ADUHELM but were not statistically significant.
In Study 1 and Study 2, ADUHELM reduced plasma p-tau 181 levelscompared to placebo. In the long-term extension of Study 1 and Study 2, a continued decrease in plasma p-Tau 181 levels was observed at the high dose through Week 128 in patients initially randomized to ADUHELM.
ADUHELM reduced CSF levels of t-Tau in substudies conducted in Study 1 and Study 2. The adjusted mean change from baseline in CSF t-Tau levels relative to placebo was in favor of the ADUHELM low (p<0.05) and high (p<0.01) dose groups at Week 78 in Study 1. Results in Study 2 numerically favored ADUHELM but were not statistically significant.
Substudies were conducted in both Study 1 and Study 2 to evaluate the effect of ADUHELM on neurofibrillary tangles composed of tau protein using PET imaging (18F-MK6240 tracer). The PET signal was quantified using the SUVR method to estimate brain levels of tau in brain regions expected to be affected by Alzheimer's disease pathology (medial temporal, temporal, frontal, cingulate, parietal, and occipital cortices) in the study population compared to a brain region expected to be spared of such pathology (cerebellum). Data from the substudies were pooled, comprising 37 patients with longitudinal follow-up. The adjusted mean change from baseline in tau PET SUVR relative to placebo at follow-up was in favor of ADUHELM high dose in the medial temporal (p<0.001), temporal (p<0.05), and frontal (p<0.05) brain regions. No statistically significant differences were observed for the cingulate, parietal, or occipital cortices.
Exposure-Response Relationships
Model based exposure-response analyses for Studies 1 and 2 demonstrated that higher exposures to ADUHELM were associated with greater reduction in clinical decline on CDR-SB, ADAS-Cog13, and ADCS-ADL-MCI. In addition, higher exposures to ADUHELM were associated with greater reduction in amyloid beta plaque in Studies 1 and 2. An association between reduction in amyloid beta plaque and clinical decline on CDR-SB was also observed.
Higher exposures to ADUHELM were associated with greater reduction in plasma p-Tau 181.
An association between reduction in plasma p-Tau 181 and reduction in Amyloid PET SUVR was observed. An association between reduction in plasma p-Tau 181 and reduced clinical decline on CDR-SB, ADAS-Cog 13, and ADCS-ADL-MCI was also observed.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of ADUHELM were characterized using a population PK analysis with concentration data collected from 2961 subjects with Alzheimer's disease who received ADUHELM in single or multiple doses.
Steady-state concentrations of ADUHELM were reached by 16 weeks of repeated dosing with an every 4-week regimen, and the systemic accumulation was 1.7-fold. The peak concentration (Cmax), trough concentration (Cmin), and area under the plasma concentration versus time curve at steady state (AUCss) of ADUHELM increased dose proportionally in the dose range of 1 to 10 mg/kg every 4 weeks.
Distribution
The mean value (95% CI) for volume of distribution at steady state is 9.63 L (9.48, 9.79).
Elimination
ADUHELM is expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG. ADUHELM clearance (95% CI) is 0.0159 (0.0156, 0.0161) L/hr. The terminal half-life is 24.8 (14.8, 37.9) days.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in other studies, including those of aducanumab-avwa or of other aducanumab products.
The immunogenicity of ADUHELM has been evaluated using an in vitro assay for the detection of binding anti-aducanumab-avwa antibodies.
In up to 41 months of treatment in the combined placebo-controlled and long-term extension periods of Studies 1 and 2, up to 0.6% (15/2689) of patients receiving ADUHELM once monthly developed anti-aducanumab-avwa antibodies.
Based on the limited number of patients who tested positive for anti-aducanumab-avwa antibodies, no observations were made concerning a potential effect of neutralizing activity of anti-aducanumab-avwa antibodies on exposure or efficacy; however, the available data are too limited to make definitive conclusions regarding an effect on pharmacokinetics, safety, or efficacy of ADUHELM. Quantification of neutralizing anti-aducanumab-avwa antibodies has not been assessed.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Impairment of Fertility
Intravenous administration of aducanumab-avwa (0, 100, 300, or 1000 mg/kg/week) to male and female rats prior to and during mating and continuing in females to gestation day 7 resulted in no adverse effects on fertility or reproductive performance.
The relevance of these data to humans is limited because aggregated amyloid beta, the pharmacological target of aducanumab-avwa, is not present in rat.
14. Clinical Studies
The efficacy of ADUHELM was evaluated in two double-blind, randomized, placebo-controlled, parallel group studies (Study 1, NCT 02484547 and Study 2, NCT 02477800) in patients with Alzheimer's disease (patients with confirmed presence of amyloid pathology and mild cognitive impairment or mild dementia stage of disease, consistent with Stage 3 and Stage 4 Alzheimer's disease, stratified to include 80% Stage 3 patients and 20% Stage 4 patients). The effects of ADUHELM were also supported by a double-blind, randomized, placebo-controlled, dose-ranging study (Study 3, NCT 01677572) in patients with Alzheimer's disease (patients with confirmed presence of amyloid pathology and prodromal or mild dementia stage of disease, consistent with Stage 3 and Stage 4 Alzheimer's disease, with an enrolled distribution of 43% Stage 3 patients and 57% Stage 4 patients), followed by an optional, dose-blind, long-term extension period.
In Studies 1 and 2, patients were randomized to receive ADUHELM low dose (3 or 6 mg/kg for ApoE ε4 carriers and noncarriers, respectively), ADUHELM high dose (10 mg/kg), or placebo every 4 weeks for 18 months, followed by an optional, dose-blind, long-term extension period. Both studies included an initial titration period of up to 6 months to the maximum target dose. At the beginning of the study, ApoE ε4 carriers were initially titrated up to a maximum of 6 mg/kg in the high dose group, which was later adjusted to 10 mg/kg.
In Studies 1 and 2, patients were enrolled with a Clinical Dementia Rating (CDR) global score of 0.5, a Repeatable Battery for Assessment of Neuropsychological Status (RBANS) delayed memory index score ≤ 85, and a Mini-Mental State Examination (MMSE) score of 24-30. In Study 3, patients were enrolled with a global CDR score of 0.5 or 1.0 and an MMSE score of 20-30. Patients were enrolled with or without concomitant approved therapies (cholinesterase inhibitors and the N-methyl-D-aspartate antagonist memantine) for Alzheimer's disease.
Studies 1 and 2 were terminated prior to their planned completion. Study endpoints were analyzed based on the prespecified statistical analysis plan.
Study 1
In Study 1, 1638 patients were randomized 1:1:1 to receive ADUHELM low dose, ADUHELM high dose, or placebo. At baseline, the mean age of patients was 71 years, with a range of 50 to 85 years.
A subgroup of 488 patients were enrolled in the amyloid PET substudy; of these, 302 were evaluated at week 78. Results from the amyloid beta PET substudy are described in Figure 1 and Table 6. CSF and plasma biomarkers are described in Table 6.
Figure 1: Reduction in Brain Amyloid Beta Plaque (Change from Baseline in Amyloid Beta PET Composite, SUVR and Centiloids) in Study 1
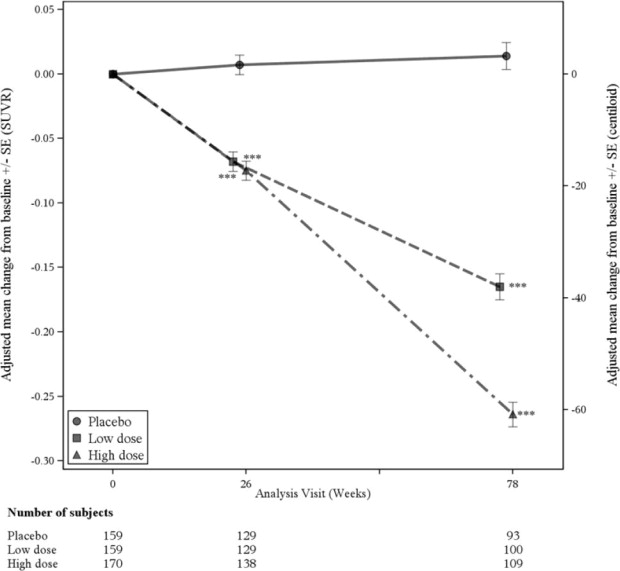
*** p<0.001
|
1 P-values were not statistically controlled for multiple comparisons. |
||
|
2 The analysis results on plasma p-Tau 181 should be interpreted with caution because of uncertainties related to stability of stored samples. |
||
| Biomarker Endpoint at Week 781 | ADUHELM
High dose | Placebo |
| Amyloid Beta PET Composite SUVR | N=170 | N=159 |
| Mean baseline | 1.383 | 1.375 |
| Change from baseline Difference from placebo | -0.264 -0.278, p<0.0001 | 0.014 |
| Amyloid Beta PET Centiloid | N=170 | N=159 |
| Mean baseline | 85.3 | 83.5 |
| Change from baseline (%) Difference from placebo | -60.8 (-71%) -64.2, p<0.0001 | 3.4 |
| CSF p-Tau 181 (pg/mL) | N=17 | N=28 |
| Mean baseline | 100.11 | 72.55 |
| Change from baseline Difference from placebo | -22.93 -22.44, p=0.0005 | -0.49 |
| Plasma p-Tau 181 (pg/mL)2 | N=294 | N=294 |
| Mean baseline | 3.342 | 3.181 |
| Change from baseline Difference from placebo | -0.424 -0.669, p<0.0001 | 0.245 |
| CSF t-Tau (pg/mL) | N=17 | N=28 |
| Mean baseline | 686.65 | 484.00 |
| Change from baseline Difference from placebo | -112.44 -112.05, p=0.0088 | -0.39 |
The primary efficacy endpoint was the change from baseline on the CDR-Sum of Boxes (CDR-SB) at Week 78. In Study 1, treatment with ADUHELM high dose demonstrated reduced clinical decline, as evidenced by a statistically significant treatment effect on change from baseline in CDR-SB compared to placebo (-0.39 [-22%], p = 0.0120), as shown in Figure 2 and Table 7. The estimate of the treatment effect favored ADUHELM across all prespecified subgroups of interest.
Figure 2: Line Plot of Primary Efficacy Endpoint (Change From Baseline in CDR Sum of Boxes) in Study 1
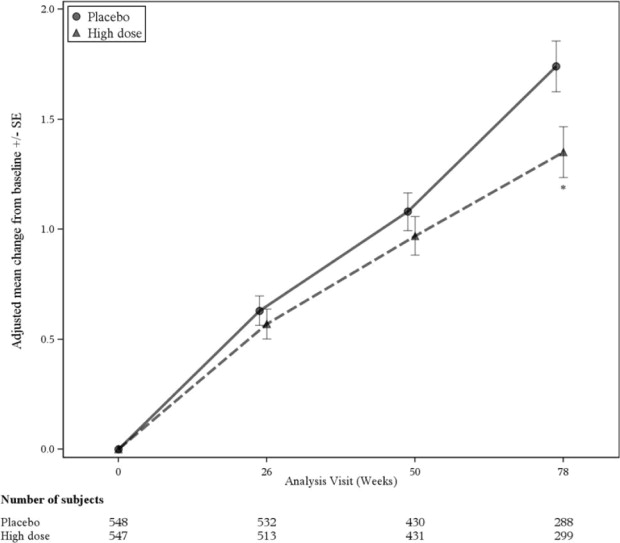
* p<0.05
Secondary efficacy endpoints included the change from baseline in MMSE score at Week 78, the change from baseline in the Alzheimer's Disease Assessment Scale-Cognitive Subscale (13 items) (ADAS-Cog 13) at Week 78, and the change from baseline in the Alzheimer's Disease Cooperative Study – Activities of Daily Living Inventory (Mild Cognitive Impairment version) (ADCS-ADL-MCI) score at Week 78. In Study 1, statistically significant differences from placebo were observed in the ADUHELM high dose group on all secondary efficacy endpoints evaluated. The estimate of the treatment effect favored ADUHELM across most prespecified subgroups of interest for the secondary efficacy endpoints. The Neuropsychiatric Inventory-10 item (NPI-10) was the only tertiary endpoint that assessed efficacy. The results of the high dose group, compared to placebo, are presented in Table 7.
Differences from placebo observed in the ADUHELM low dose group numerically favored ADUHELM but were not statistically significant.
|
1P-value was not statistically controlled for multiple comparisons. |
||
| Clinical Endpoint at Week 78 | ADUHELM High dose
(N=547) | Placebo
(N=548) |
| CDR-SB | ||
| Mean baseline | 2.51 | 2.47 |
| Change from baseline Difference from placebo (%) | 1.35 -0.39 (-22%) p=0.0120 | 1.74 |
| MMSE | ||
| Mean baseline | 26.3 | 26.4 |
| Change from baseline Difference from placebo (%) | -2.7 0.6 (-18%) p=0.0493 | -3.3 |
| ADAS-Cog 13 | ||
| Mean baseline | 22.246 | 21.867 |
| Change from baseline Difference from placebo (%) | 3.763 -1.400 (-27%) p=0.0097 | 5.162 |
| ADCS-ADL-MCI | ||
| Mean baseline | 42.5 | 42.6 |
| Change from baseline Difference from placebo (%) | -2.5 1.7 (-40%) p=0.0006 | -4.3 |
| NPI-101 | ||
| Mean baseline | 4.5 | 4.3 |
| Change from baseline Difference from placebo (%) | 0.2 -1.3 (-87%) p=0.0215 | 1.5 |
Study 2
In Study 2, 1647 patients were randomized 1:1:1 to receive ADUHELM low dose, ADUHELM high dose, or placebo. At baseline, the mean age of patients was 71 years, with a range of 50 to 85 years.
A subgroup of 585 patients were enrolled in the amyloid PET subgroup; of these, 374 were evaluated at week 78. Results from the amyloid beta PET substudy are described in Figure 3 and Table 8. CSF and plasma biomarkers are described in Table 8.
Figure 3: Reduction in Brain Amyloid Beta Plaque (Change from Baseline in Amyloid Beta PET Composite, SUVR and Centiloids) in Study 2
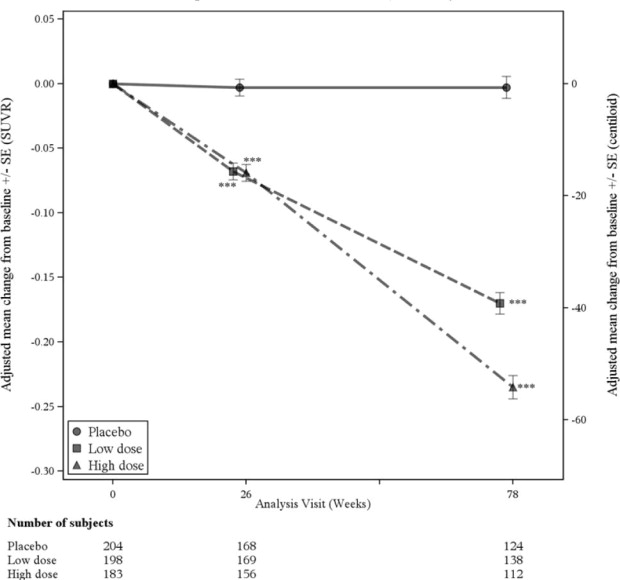
*** p<0.001
|
1 P-values were not statistically controlled for multiple comparisons. |
||
|
2 The analysis results on plasma p-Tau 181 should be interpreted with caution because of uncertainties related to stability of stored samples. |
||
| Biomarker Endpoint at Week 781 | ADUHELM
High dose | Placebo |
| Amyloid Beta PET Composite SUVR | N=183 | N=204 |
| Mean baseline | 1.407 | 1.376 |
| Change from baseline Difference from placebo | -0.235 -0.232, p<0.0001 | -0.003 |
| Amyloid Beta PET Centiloid | N=183 | N=204 |
| Mean baseline | 90.8 | 83.8 |
| Change from baseline (%) Difference from placebo | -54.0 (-59%) -53.5, p<0.0001 | -0.5 |
| CSF p-Tau 181 (pg/mL) | N=18 | N=15 |
| Mean baseline | 121.81 | 94.53 |
| Change from baseline Difference from placebo | -13.19 -10.95, p=0.3019 | -2.24 |
| Plasma p-Tau 181 (pg/mL)2 | N = 285 | N = 334 |
| Mean baseline | 3.131 | 3.180 |
| Change from baseline Difference from placebo | -0.484 -.0769, p<0.0001 | 0.286 |
| CSF t-Tau (pg/mL) | N=16 | N=14 |
| Mean baseline | 618.50 | 592.57 |
| Change from baseline Difference from placebo | -102.51 -69.25, p=0.3098 | -33.26 |
No statistically significant differences were observed between the ADUHELM-treated and placebo-treated patients on the primary efficacy endpoint, the change from baseline in CDR-SB score at 78 weeks.
Study 3
In Study 3, 197 patients were randomized to receive a fixed dose of ADUHELM 1 mg/kg (n=31), 3 mg/kg (n=32), 6 mg/kg (n=30), 10 mg/kg (n=32), titration of ADUHELM to 10 mg/kg over 44 weeks (n=23), or placebo (n=48) for 12 months. At baseline, the mean age of patients was 73 years, with a range of 51-91 years.
Results from the amyloid beta PET substudy are described in Figure 4 and Table 9.
Figure 4: Reduction in Brain Amyloid Beta Plaque (Change from Baseline in Amyloid Beta PET Composite, SUVR and Centiloids ) in Study 3
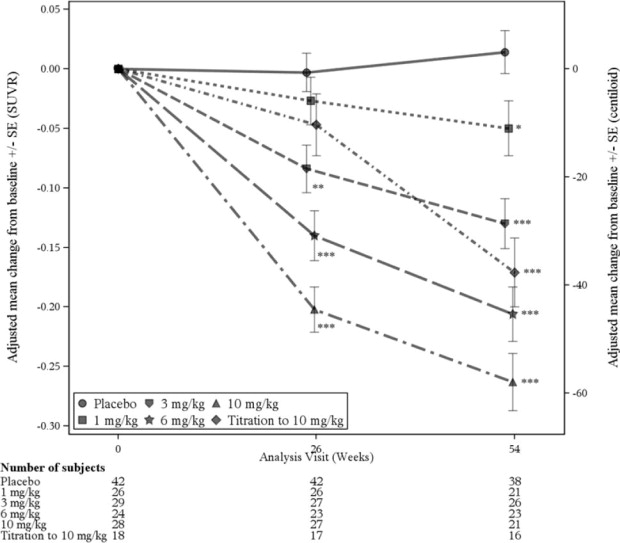
* p<0.05, ** p<0.01, *** p<0.001
|
1P-values were not statistically controlled for multiple comparisons. |
||
| Biomarker Endpoint at Week 541 | ADUHELM
10 mg/kg | Placebo |
| Amyloid Beta PET Composite SUVR | N=28 | N=42 |
| Mean baseline | 1.432 | 1.441 |
| Change from baseline Difference from placebo | -0.263 -0.277, p<0.0001 | 0.014 |
| Amyloid Beta PET Centiloid | N=28 | N=42 |
| Mean baseline | 94.5 | 96.5 |
| Change from baseline (%) Difference from placebo | -58.0 (-61%) -61.1, p<0.0001 | 3.1 |
Clinical assessments in Study 3 were exploratory. Results for clinical assessments were directionally aligned with the findings from Study 1, with less change from baseline in CDR-SB and MMSE scores at 1 year in the ADUHELM 10 mg/kg fixed-dose group than in patients on placebo (CDR-SB: -1.26, 95% CI [-2.356, -0.163]; MMSE: 1.9, 95% CI [0.06, 3.75]).
16. How is Aduhelm supplied
16.1 How Supplied
ADUHELM (aducanumab-avwa) injection is a preservative-free, sterile, clear to opalescent, and colorless to yellow solution. ADUHELM is supplied one vial per carton as follows:
170 mg/1.7 mL (100 mg/mL) single-dose vial (with red flip cap) – NDC 64406-101-01
300 mg/3 mL (100 mg/mL) single-dose vial (with blue flip cap) – NDC 64406-102-02
16.2 Storage and Handling
Unopened Vial
- Store in original carton until use to protect from light.
- Store in a refrigerator at 2°C to 8°C (36°F to 46°F).
- not freeze or shake.
- If no refrigeration is available, ADUHELM may be stored unopened in its original carton to protect from light at room temperature up to 25°C (77°F) for up to 3 days.
- Prior to dilution, unopened vials of ADUHELM may be removed from and returned to the refrigerator if necessary, when kept in the original carton. Total combined time out of refrigeration with protection from light should not exceed 24 hours at room temperature up to 25°C (77°F).
17. Patient Counseling Information
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Medication Guide).
Amyloid Related Imaging Abnormalities
Inform patients that ADUHELM may cause Amyloid Related Imaging Abnormalities or “ARIA”. ARIA most commonly presents as temporary swelling in areas of the brain that usually resolves over time. Some people may also have small spots of bleeding in or on the surface of the brain. Inform patients that most people with swelling in areas of the brain do not experience symptoms, however, some people may experience symptoms such as headache, confusion, dizziness, vision changes, nausea, aphasia, weakness, or seizure. Instruct patients to notify their healthcare provider if these symptoms occur. Inform patients that events of intracerebral hemorrhage greater than 1 cm in diameter have been reported infrequently in patients taking ADUHELM, and that the use of anticoagulant or thrombolytic medications while taking ADUHELM may increase the risk of bleeding in the brain. Notify patients that their healthcare provider will perform MRI scans to monitor for ARIA [see Warnings and Precautions (5.1)].
Inform patients that although ARIA can occur in any patient treated with ADUHELM, there is an increased risk in patients who are ApoE ε4 homozygotes, and that testing for ApoE ε4 status should be performed prior to initiation of treatment to inform the risk of developing ARIA. Prior to testing, discuss with patients the risk of ARIA across genotypes and the implications of genetic testing results. Inform patients that if testing is not performed, it cannot be determined if they are ApoE ε4 homozygotes and at a higher risk for ARIA.
Patient Registry
Advise patients that the Alzheimer's Network for Treatment and Diagnostics (ALZ-NET) is a voluntary provider-enrolled patient registry that collects information on treatments for Alzheimer's disease, including ADUHELM. Encourage patients to participate in the ALZ-NET registry [see Warnings and Precautions (5.1)].
Hypersensitivity Reactions
Inform patients that ADUHELM may cause hypersensitivity reactions, including angioedema and urticaria, and to contact their healthcare provider if hypersensitivity reactions occur [see Warnings and Precautions (5.2)].
55093-06
Manufactured by:
Biogen Inc.
Cambridge, MA 02142
US License #1697
ADUHELM is a registered trademark of Biogen.
© 2023 Biogen
|
This Medication Guide has been approved by the U.S. Food and Drug Administration |
Revised: 08/2023 |
||
| MEDICATION GUIDE
ADUHELM® (AD-yew-helm) (aducanumab-avwa) injection, for intravenous use |
|||
|
What is the most important information I should know about ADUHELM?
|
|||
|
| ||
| Some people have a genetic risk factor (homozygous apolipoprotein E gene carriers) that may cause an increased risk for ARIA. Talk to your healthcare provider about testing to see if you have this risk factor. Some medicines may increase the risk for larger areas of bleeding in the brain in patients taking ADUHELM. Talk to your healthcare provider to see if you are on any medicines that increase this risk. Your healthcare provider will do magnetic resonance imaging (MRI) scans before and during your treatment with ADUHELM to check you for ARIA. Call your healthcare provider or go to the nearest hospital emergency room right away if you have any of the symptoms listed above. |
|||
| What is ADUHELM?
ADUHELM is a prescription medicine used to treat people with Alzheimer's disease. It is not known if ADUHELM is safe and effective in children. |
|||
Before receiving ADUHELM, tell your healthcare provider about all of your medical conditions, including if you:
|
|||
| Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Especially tell your healthcare provider if you take medicines to reduce blood clots from forming (antithrombotic medicines, including aspirin). Ask your healthcare provider for a list of these medicines if you are not sure. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacists when you get a new medicine. |
|||
How will I receive ADUHELM?
|
|||
| What are the possible side effects of ADUHELM?
ADUHELM can cause serious side effects, including:
|
|||
The most common side effects of ADUHELM include:
|
|||
| These are not all the possible side effects of ADUHELM. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
| General Information about the safe and effective use of ADUHELM.
Medicines are sometimes prescribed for purposes other than those listed in this Medication Guide. You can ask your pharmacist or healthcare provider for more information about ADUHELM that is written for health professionals. There is a registry that collects information on treatments for Alzheimer's disease. The registry is named ALZ-NET (Alzheimer's Network for Treatment and Diagnostics). Your healthcare provider can help you become enrolled in this registry. For more information, go to www.aduhelm.com or call at 1-833-425-9360. |
|||
| What are the ingredients in ADUHELM?
Active ingredient: aducanumab-avwa Inactive ingredients: L-arginine hydrochloride, L-histidine, L-histidine hydrochloride monohydrate, L-methionine, polysorbate 80, and water for injection Manufactured by: Biogen Inc., Cambridge, MA 02142, U.S. License #1697 ADUHELM is a registered trademark of Biogen. ©2023 Biogen |
|||

| ADUHELM
aducanumab injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ADUHELM
aducanumab injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Biogen Inc. (121376230) |
More about Aduhelm (aducanumab)
- Check interactions
- Compare alternatives
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: miscellaneous central nervous system agents
- Breastfeeding
- En español